E.V. Tozliyan, endocrinolog pediatru, genetician, dr., I.V. Shulyakova, neurolog, dr.,
izolat subdiviziune structurală„Institutul Clinic de Cercetare de Pediatrie” SBEE HPE „Universitatea Națională de Cercetare Medicală din Rusia numită după N.I. Pirogov” de la Ministerul Sănătății al Federației Ruse, Moscova
Cuvinte cheie:
copii, pseudohipoparatiroidism, osteodistrofie ereditară Albright, obezitate, hipocalcemie, diagnostic, rezistență la hormonul paratiroidian.
Cuvinte cheie: copii, pseudohipoparatiroidism, osteodistrofie ereditară Albright, obezitate, hipocalcemie, diagnostic, rezistență la hormoni paratiroidieni.
Pseudohipoparatiroidism (pseude greacă - fals + hipoparatiroidism; sinonim: osteodistrofia ereditară a lui Albright, sindromul „pui javanez”) - o boală ereditară rară sistemul osos, care imită hipoparatiroidismul și se caracterizează printr-un metabolism afectat de calciu și fosfor; adesea însoţită de retard mintal şi dezvoltarea fizică. Boala a fost descrisă pentru prima dată de endocrinologul american Albright F. în 1942. Prevalența bolii este de 7,9 la 1 milion de oameni.
DATE GENETICE
Pseudohipoparatiroidismul (PHP) este o boală eterogenă genetic. Datele despre tipul de transmitere ereditară sunt contradictorii: atât tip dominant X-linked, cât și autozomal dominant, autozomal recesiv. În cele mai multe cazuri, dezvoltarea osteodistrofiei ereditare a lui Albright este asociată cu mutații în locusul 20q13 al genei GNAS1 situată pe cromozomul 20 (Patten și colab., 1990), care codifică proteina Gs-alfa asociată cu hormonul paratiroidian (PTH) receptor. Un fenotip similar a fost găsit și la pacienții cu deleție interstițială a brațului lung al cromozomului 2 al locusului 2q37.
PATOGENEZĂ
Patogenia pseudohipoparatiroidismului se bazează pe rezistența determinată genetic a rinichilor și a scheletului la acțiunea parathormonului ca urmare a unui defect al complexului „citoreceptor specific - hormon paratiroidian - adenilat ciclază”, care perturbă formarea ciclicului 3 "- , 5"-adenozin monofosfat (cAMP) în rinichi, care este mediatorul intracelular al acțiunii hormonului paratiroidian asupra proceselor metabolice. Pseudohipoparatiroidismul este genetic boala eterogena. La unii pacienți, citoreceptorul în sine este defect, care leagă hormonul paratiroidian (pseudohipoparatiroidism de tip 1A), la alții există un defect în proteina de legare a nucleotidelor localizată în stratul dublu lipidic al membranei celulare și leagă funcțional receptorul de adenilat ciclază ( pseudohipoparatiroidism de tip 1B). Unii pacienți au deficit enzimatic al adenilat-ciclazei în sine (pseudohipoparatiroidism tip 2). Deficiența cAMP, rezultată din aceste defecte, duce la o încălcare a sintezei proteinelor specifice care determină efectul biologic al hormonului paratiroidian. Astfel, se pierde sensibilitatea organelor țintă la hormonul paratiroidian.
CARACTERISTICI CLINICE
În prezent sunt 4 forme clinice patologii: tipurile 1A, 1B, 1C și 2. Cunoașterea caracteristicilor și datelor clinice și biochimice ale acestora cercetare genetică permite diagnosticul diferențial în cadrul formei nosologice în sine.
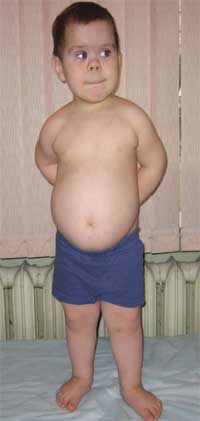
Semnele comune care fac posibilă suspectarea bolii sunt dezvoltarea fizică disproporționată, statura mică (până la nanism) din cauza scurtării. extremitati mai joase(foto 1), brahidactilie (foto 2), față rotundă „în formă de lună” (foto 3). Uneori apar exostoze și aplazie a dinților.
Fotografie 1.
Aspect copil cu osteodistrofie Albright
(trăsături ale fenotipului, statură mică din cauza scurtării membrelor inferioare)
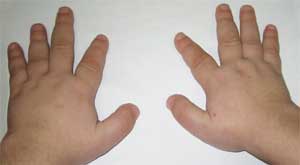
Poza 2.
Caracteristicile sistemului osos la pacient
cu osteodistrofie Albright
(brahidactilie - degete scurte)
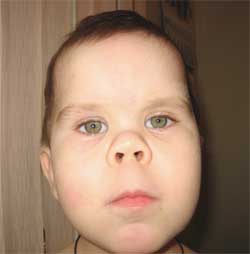
Poza 3.
Caracteristicile fenotipului copilului
cu osteodistrofie Albright
(fața rotundă de „lună”)
O scurtare accentuată a oaselor metacarpiene și metatarsiene I, III și V (în special III și IV) este considerată un semn patognomonic, drept urmare degetele II de pe mâini și picioare sunt mai lungi decât celelalte, iar când mâna este strâns într-un pumn, nu există umflături în regiunea articulațiilor metacarpofalangiene IV și V - așa-numitul brahimetafalangism. De asemenea, sunt detectate falange scurte late, ingrosarea boltii craniene si demineralizarea oaselor (osteoporoza), obezitatea.
Retardarea mintală (adesea de severitate moderată) se găsește la aproximativ 20% dintre pacienți. După unii autori, oligofrenia apare în 70% din cazuri cu hipocalcemie și în 30% din cazuri cu normocalcemie. procesele mentale pacientii sunt lente. În starea neurologică se remarcă adesea stinghereală motrică, reacții nevrotice: temeri, anxietate, neliniște, vis urât, reflexe crescute, convulsii de natura tetanica si cauzate de hipocalcemie, paroxisme uneori convulsive. Sunt descrise și simptome miopatice: oboseală musculară, slăbiciune musculară. Se observă adesea tulburări extrapiramidale: hiperkineză coreiformă, atetoză, hemispasm facial, parkinsonism, în unele cazuri apar paroxisme epileptice, simptome cerebeloase: ataxie, tulburări de coordonare.
Adesea, se determină calcificări ale țesuturilor moi, calcificări subcutanate (piept, abdomen, tendoane calcaneane), cu examen histologic din care - osteom cutis(Izraeli et al., 1992), creier (ganglionii bazali). Este important de reținut că calcificări pot fi deja prezente la naștere. Din cauza hipocalcemiei, de obicei se dezvoltă cataracta și apar defecte ale smalțului dentar.
PSEUDOHIPOPARATIROIZA TIP 1A
are un mod de moștenire autosomal dominant. Gena pseudohipoparatiroidismului de tip 1A, GNAS1, este localizată pe brațul lung al cromozomului 20, la locusul 20q13.2. Dezvoltarea bolii este asociată cu o deficiență a proteinei de legare a guaninei-nucleotide (proteina Gs). În același timp, PTH, legându-se de receptorii țesuturilor țintă, nu este capabil să activeze adenozin monofosfatul ciclic (cAMP) și să provoace un răspuns tisular. Probabil, un mecanism similar stă la baza dezvoltării insensibilității țesuturilor altor organe și glandelor endocrine (hipofuncție glanda tiroida, gonade, glanda pituitară, diabet zaharat, precum și un răspuns hepatic redus la administrarea de glucagon), observat în pseudohipoparatiroidismul tip 1A. Cu acest tip de patologie, nu există o excreție crescută de AMPc în urină, care este caracteristică normei, ca răspuns la administrarea exogenă de PTH. Boala este diagnosticată mai des la vârsta de 5-10 ani. Pacienții au statură mică, gâtul scurt, fața rotundă, scurtarea oaselor metacarpiene și metatarsiene (mai des scurtarea degetelor IV și mai rar II) - așa-numitul brahi-metafalangism. Se notează calcificarea țesuturilor moi, calcificări subcutanate, care pot fi detectate deja la naștere; adesea apare o implicare simultană a altor glande endocrine: glanda tiroidă (hipofuncție), gonade, pancreas (diabet zaharat). Din cauza hipocalcemiei, se dezvoltă adesea cataractă și un defect al smalțului dentar. Ca test de diagnostic diferențial pentru diferența dintre PGP tip 1A și hipoparatiroidism: absența unui efect clinic din administrarea parenterală a PTH sub forma unei creșteri a nivelului de calciu în sânge și a unei creșteri a excreției renale a fosforului în urina (efect fosfaturic).
Examenul biochimic relevă hipocalcemie, hiperfosfatemie, o creștere a nivelului de hormon paratiroidian în sânge și hipofosfaturie. Nivelul proteinei Gs din sânge este redus. La examinare cu raze X ale sistemului osos se constată scurtarea oaselor metacarpiene și metatarsiene, demineralizare generalizată, îngroșarea oaselor bolții craniene.
PSEUDOHIPOPARATIROIZA TIP 1B
are un tip de moștenire autosomal dominant, cu toate acestea, un tip de moștenire dominant, legat de X, nu este exclus. Este necesar să se țină seama de penetrarea incompletă uneori observată a genei bolii și de posibilitatea unui transport ascuns al patologiei. Prin urmare, se recomandă examinarea clinică (depistarea evoluției subclinice a bolii) și biochimică (determinarea nivelurilor de calciu, fosfor, PTH din sânge) ale presupușilor purtători ai bolii. HPT tip 1B este cauzat de o deficiență a receptorilor tisulari pentru hormonul paratiroidian în organele țintă și rezistența limitată la hormonul paratiroidian. Tabloul clinic este similar cu cel al clinicii de tip 1A, dar nu există leziuni ale altor glande endocrine, iar osteodistrofia este mai puțin frecventă.
La pacienți, nu există o reacție renală la administrarea exogenă a hormonului paratiroidian sub forma unei creșteri a excreției de adenozin monofosfat ciclic în urină, cu toate acestea, spre deosebire de tipul 1A, nivelul proteinei Gs din sânge este normal. Femeile sunt afectate mai des decât bărbații, dar severitatea bolii poate fi aceeași atât la bărbați, cât și la femei.
PSEUDOHIPOPARATIROIZA TIP 1C
unii autori se identifică cu pseudo-pseudohipoparatiroidismul (PPHP) descris de Albright F. în 1952. Se caracterizează printr-un tablou clinic caracteristic HAP, dar nivelurile de calciu, fosfor din sânge și urină rămân în limitele normale. Indicatorii PTH și proteinei Gs din sânge sunt, de asemenea, stocați nivel normal. Unii pacienți cu PHP tip 1C au deleții de novo pe cromozomul 2. Este posibil ca această variantă a bolii să fie un subtip de HPP tip 1A.
PSEUDOHIPOPARATIROZĂ TIP 2
similar clinic cu alte tipuri de boală, dar are un mod de moștenire autosomal recesiv. Nu este exclusă existența formelor de patologie autosomal dominante. Patogenia dezvoltării este asociată cu rezistența intracelulară la cAMP. În acest caz, PTH se leagă de receptori și provoacă un răspuns celular normal la PTH sub forma unei creșteri a excreției de cAMP. Cu toate acestea, insensibilitatea intracelulară la AMPc nu permite realizarea deplină a acțiunii PTH. În același timp, reacția normală a rinichilor la administrarea exogenă a hormonului paratiroidian este păstrată sub forma unei creșteri a excreției de adenozin monofosfat ciclic în urină. S-a sugerat că tipul 2 HWP poate fi asociat cu deficiența de vitamina D.
Astfel, tipurile de PHP identificate sunt caracterizate clinic prin sensibilitatea redusă a organelor țintă la PTH, dar diferă în mecanismele patogenetice de formare a insensibilității tisulare.
DIAGNOSTICĂ
Natura excreției renale de AMPc ca răspuns la administrarea de PTH poate servi ca test de diagnostic diferențial de laborator: excreția crescută de AMPc este observată în tipul 2 și absența acesteia în tipul 1. Diagnosticul este confirmat prin detectarea unui nivel redus de guanină-nucleotidă. -proteina de legare (proteina Gs) in sange (in medie de 1,5-2 ori) fata de norma. Hipocalcemia este de obicei asociată cu hiperfosfatemie și hipofosfaturie. Nivelul de PTH este crescut; cu tipul 1C, nivelul PTH este normal, ceea ce a dat naștere denumirii de „pseudohipoparatiroidism”. Examinarea cu raze X a sistemului osos relevă scurtarea oaselor metacarpiene și metatarsiene, adesea demineralizare generalizată (osteoporoză), îngroșarea oaselor bolții craniene. Desenul dermatoglific prezintă o deplasare a triradiusului palmar axial.
Criterii de diagnostic:
- creștere scăzută;
- fata rotunda;
- întârziere a nervos dezvoltare mentală;
- anomalii ale scheletului;
- calciu seric scăzut;
- niveluri ridicate de hormon paratiroidian în sânge;
- scăderea excreției urinare de fosfat și cAMP.
TRATAMENT ŞI PREVENIRE
Tratamentul hipocalcemiei constă în administrarea de suplimente de calciu în doze suficiente pentru a menține o concentrație normală de calciu în sânge. Mare importanță are terapie cu vitamina D. În prezent, se folosesc metaboliți activi de vitamina D - oksidevit, 1-alfa-D3, calcitrină etc. în doză de 1-2 mcg/zi cu rezultat pozitiv (creșterea calciului în sânge, reducerea manifestărilor de sindrom convulsiv). Tachistin (0,5–1,5 mg/zi) este de asemenea eficient. Acest medicament crește absorbția calciului în intestine și, prin urmare, crește nivelul de calciu din sânge. Terapia anticonvulsivante este utilizată ca tratament suplimentar. Tratamentul nu are un efect vizibil asupra dezvoltării intelectuale, dar împreună cu o scădere a simptomelor sindromului convulsiv se observă o regresie a manifestărilor neurologice (tulburări subcorticale, hiperkinezie coreiformă, atetoză etc.). Pentru a evita supradozajul preparatelor cu vitamina D, este necesar să se controleze concentrația de calciu din sânge la fiecare 3-7 zile în primele 2 săptămâni de tratament și în fiecare lună în următoarele 2-3 luni. La atingerea unei concentrații stabile de calciu în sânge, este suficient să o verifici o dată la 2-3 luni. O dietă cu restricții în fosfor ajută la normalizarea atât a concentrației de fosfor, cât și de calciu în sânge și la eliminarea simptomelor hiperparatiroidismului secundar. În caz de insuficiență a altor glande endocrine, terapie de substituție hormonii corespunzători.
Tratamentul cu hormon paratiroidian este ineficient. Pentru ameliorarea crizelor convulsive se administrează intravenos o soluție 10% de clorură de calciu sau gluconat de calciu; în interior - 5-10% soluție de clorură de calciu, 1 lingură de 3-4 ori pe zi: gluconat de calciu, lactat de calciu - până la 10 g pe zi.
PROGNOZA căci viaţa este determinată de severitatea sindromului convulsiv.
PREVENIRE boala se bazează pe datele consilierii genetice medicale.
CONSILIERE MEDICAL GENETICĂ
În consilierea genetică medicală, trebuie să se procedeze de la tipul de moștenire autozomal dominant și un risc ridicat (50%) de reapariție a bolii în familia cu forme moștenite. Pentru a identifica natura tipului de moștenire, este necesar să se efectueze o examinare amănunțită a părinților, deoarece sindromul se poate manifesta cu simptome clinice minime. În prezent, diagnosticul genetic molecular al bolii a fost dezvoltat și este îmbunătățit prin tiparea mutațiilor în gena GNAS1 de pe cromozomul 20. Sunt în curs de dezvoltare metode de diagnosticare prenatală a bolii în ansamblu și a tipurilor sale individuale.
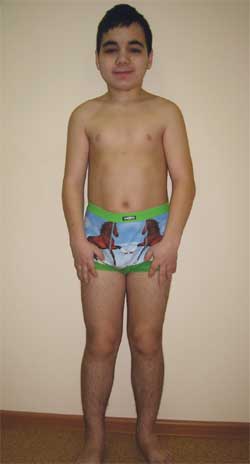
OBSERVAȚIE CLINICĂ Băiatul G., 14,5 ani (foto 4), a fost internat la Institutul de Cercetare Clinică de Pediatrie cu diagnostic de boală degenerativă sistem nervos? hidrocefalie externă congenitală; epilepsie simptomatică; sindrom ereditar? boala de depozitare? encefalopatie metabolică; hipotiroidism subclinic; statură mică de geneză mixtă; tulburari cognitive.
Reclamații la internarea la dureri de cap paroxistice intense localizate în zona frontalași însoțite de vărsături, care aduce alinare, scăderea memoriei și a performanței școlare, atacuri convulsive, în timpul cărora apar zvâcniri la mâna dreaptă.
Fotografie 4.
Copil G., 14,5 ani, cu osteodistrofie Albright
(caracteristici ale fenotipului, statură mică, scurtarea membrelor, brahidactilie)
Istorie de familie: Părinții sunt armeni după naționalitate, nu sunt rudă de sânge și nu au riscuri profesionale. În pedigree-ul cazurilor boală mintală, epilepsie, întârzieri de dezvoltare nu au fost observate. Frați, soră, 17 ani - din cuvinte - sănătos.
Istoria vieții și a bolii: un băiat din a 2-a sarcină, care a decurs fără trăsături, a doua naștere, la timp, fiziologic, greutate la naștere - 3100 g, lungime - 51 cm. A țipat imediat, scor Apgar - 7/9 puncte. Deteriorarea in ziua a 3-a - convulsii neonatale, oprite in maternitate. Perioada postnatală timpurie - fără caracteristici. A existat o ușoară întârziere a tempo-ului în dezvoltarea motrică în primul an de viață, mers independent de la 1 an și 3 luni. În acest sens, el a fost observat de un neurolog cu un diagnostic de afectare organică a sistemului nervos central; hidrocefalie congenitală; convulsii neonatale; convulsii febrile în istorie.
A primit diakarb, finlepsină. Debutul crizelor de la 1 an 11 luni. – asimetric, tonic sub formă de tensiune mana dreapta si picioare, cu stabilirea ochilor, pana la 2 minute, fara pierderea cunostintei, frecvente pana la 10 episoade pe zi. A primit depakine neregulat. Pe fondul auto-anulării - un singur statut tonic. La vârsta de 2 ani a fost efectuată o scanare CT a creierului la locul de reședință, unde au fost detectate un singur focar de demielinizare în lobii occipitali.
Consultat de un neurochirurg, tratament conservator recomandat. De la varsta de 3 ani apare o intarziere in dezvoltarea psihoverbala, se recomanda supravegherea unui psihiatru.
De la vârsta de 4-5 ani, părinții au început să observe deformarea și scurtarea degetelor de la picioare și mâinilor, în special degetele II-IV simetric pe mâini și picioare, și o scădere a ratei de creștere. La vârsta de 8 ani, concluzia unui logoped - încălcarea generală vorbire de nivelul 2-3, antrenament în scoala de specialitate. La aceeași vârstă, examinare de către un genetician la locul de reședință, concluzie: boala ereditara schimb valutar? s-a recomandat un studiu al aminoacizilor din sânge, nu au fost detectate modificări; concluzie finală: nu au fost dezvăluite date pentru o boală metabolică ereditară; hipocondroplazie; tratament recomandat de un neurolog și endocrinolog.
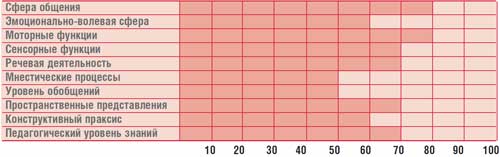
Masa.
Profilul dezvoltării mentale a copilului G., 14,5 ani (IQ = 68)
La vârsta de 8 ani, a fost consultat de un endocrinolog despre întârzierea creșterii și dezvoltării. La examinarea cu raze X a mâinilor, s-au observat următoarele caracteristici: falangele mijlocii, principale și oasele metacarpiene au fost scurtate și îngroșate; Diagnosticul radiologului a fost acondroplazie.
Examinat în mod repetat la locul de reședință într-un spital neurologic. La vârsta de 12 ani au apărut crize convulsive fără pierderea cunoștinței cu zvâcniri ale mâinii drepte, care sunt de natură în serie, a fost prescrisă terapia anticonvulsivante (Depakine), frecvența convulsiilor a scăzut semnificativ. La vârsta de 13 ani, a fost efectuat un RMN al creierului cu contrast - modificări simetrice în baza lobilor temporali la nivelul nucleelor sub forma unei creșteri a semnalului RM, care este tipic pentru toxic (mangan). sau encefalopatii metabolice (cupru, fier).
Reexaminat la vârsta de 13 ani 3 luni. un endocrinolog, în studiul profilului tiroidian a fost detectată o creștere a hormonului de stimulare a tiroidei (TSH), a fost diagnosticată hipotiroidismul subclinic și a fost prescrisă L-tiroxină.
La analizarea cardului de ambulatoriu al copilului și a documentației la locul de reședință, studiul calciului și fosforului a fost efectuat o dată, la vârsta de 1,5 ani, s-a notat hipocalcemie, dar această ocazie examinarea de urmărire nu a fost efectuată. Având în vedere incertitudinea diagnosticului la locul de reședință, geneticianul a trimis copilul la Moscova, la Institutul Clinic de Cercetare Științifică de Pediatrie, pentru a clarifica diagnosticul.
Date obiective ale cercetării:
Înălțime - 143 cm, greutate - 43 kg.
Dezvoltarea fizică este foarte scăzută, armonioasă, fizicul este disproporționat din cauza scurtării membrelor. Sds de creștere corespunde la -2,8 abateri de la normă (normal -2 + 2).
Caracteristici fenotipului: față rotundă, gât scurt, incizie antimongoloidă a fisurilor palpebrale, punte lată a nasului, frunte înaltă, brahidactilie, scurtarea oaselor metacarpiene și metatarsiene IV și V (foto 5). Pe organele interne - fără caracteristici. Dezvoltare sexuală – Stadiul Tanner III-IV (care corespunde vârstei).
Date din studii de laborator și funcționale:
Analiza clinica sângele și urina sunt normale.
Test de sânge biochimic: calciu total - 1,39 (normal 2,02-2,6 mmol / l), calciu ionizat - 0,61 (normal 1,13-1,32 mmol / l), fosfor anorganic - 3,66 (normă 0,86-1,56 mmol / l), alți indicatori sunt în limita intervalul normal.
Analiza biochimică a urinei: excreția renală de fosfați este redusă - 11,5 mmol / l (normă 19-32 mmol / l).
Profil tiroidian: TSH - 11,75 (normă 0,4-4,0 μIU/ml), T4 liber - 0,49 (normă 1,0-1,8 ng/dl).
Hormon paratiroidian - 499 (normal 12-65 pg / ml), hormon de creștere - 7 ng / ml (normal 7-10 ng / ml), somatomedin-C - 250 ng / ml (normal 88-360 ng / ml).
ecografie organe interne- fără caracteristici.
ECG - migrarea stimulatorului cardiac supraventricular pe fondul unei frecvențe cardiace regulate de 71-80 bătăi/min. Blocarea incompletă a piciorului drept al mănunchiului lui His. Încălcarea procesului de repolarizare la nivelul miocardului peretele din spate ventriculul stâng (scăderea z.T III, aVF).
R-grafia coloanei vertebrale - scolioza dreapta toracic coloana vertebrală de gradul I, osteoporoză severă.
R-grafie a mâinilor cu captarea antebrațelor - scurtarea și extinderea falangelor terminale și mijlocii. Vârsta osoasă - 13,5–14 ani.
Modelele EEG ale activității epileptice nu au fost înregistrate.
RMN al creierului - imagine RMN a focarelor subcorticale multiple de semnal MR crescut în Lobii frontali, hidrocefalie externă compensată cu atrofie a substanței cerebrale.
MSCT a creierului - zone simetrice de calcificare a nucleilor lentiformi. Zone hiperdense difuze în talamus, nuclei caudați cu o zonă de calcificare în dreapta. Multiple calcificări punctate ale țesuturilor moi tegumentare ale craniului.
Audiograma - fără patologie.
Diagnosticarea ADN-ului în gena GNAS1 este în curs.
Consultanță de specialitate:
Endocrinolog - osteodistrofia ereditară a lui Albright tip 1A (pseudohipoparatiroidism). Hipotiroidismul primar, compensație medicală incompletă.
Oftalmolog - cataractă secundară completă. Recomandat tratament chirurgical.
Psiholog - tulburări cognitive (profilul psihologic al copilului este prezentat în tabel).
Luând în considerare fenotipul copilului, datele anamnezei, rezultatele cercetări suplimentare(hipocalcemie, hiperfosfatemie, hipofosfaturie, creșterea hormonului paratiroidian sanguin), calcificări în substanța creierului, prezența cataractei, hipotiroidie), s-a pus un diagnostic: osteodistrofie ereditară Albright tip 1A (pseudohipoparatiroidism). Se recomandă efectuarea diagnosticului ADN - căutarea mutațiilor în gena GNAS1.
Tratament: se recomandă copilului să ia eutirox în doză de 100 mcg/zi; metabolit activ al vitaminei D - alfa-D3 ("Teva") în doză de 2 mcg/zi; calciu ("Sandoz") 2000 mg/zi; aport constant de terapie anticonvulsivante - finlepsin 800 mg / zi sub supravegherea unui neurolog-epileptolog; cursuri cu un logoped-defectolog și un psiholog; terapie energotropă (Elkar și coenzima Q10 în doze de vârstă). Controlul indicatorilor metabolismului fosfor-calciu, nivelurile hormonilor paratiroidieni.
Prin urmare, Observația clinică prezentată demonstrează complexitatea căutării diagnosticului diferențial, importanța studierii în timp util a parametrilor biochimici simpli (în caz de epilepsie, screening-ul repetat al indicatorilor metabolismului fosfor-calciu este obligatoriu), rezultatele diagnosticării tardive a unei boli determinate genetic. , necesitatea de a integra semne individuale în fenotipul general al unei anumite stări patologice pentru diagnosticarea în timp util a anumitor forme de boli ereditare. Diagnosticul în timp util, clarificarea genezei fiecărui sindrom sunt deosebit de importante, deoarece vă permit să găsiți cea mai bună abordare pentru tratamentul acestor afecțiuni, prevenire posibile complicații(până la handicapul copilului); prevenirea recidivei bolilor ereditare în familiile afectate (consiliere genetică medicală). Acest lucru dictează necesitatea medicilor de diferite specialități de a naviga clar în fluxul patologiei ereditare. Bibliografia este în curs de revizuire.
Pseudohipoparatiroidism - suficient boala rara sistemul osos, a cărui esență este o încălcare a schimbului de calciu și fosfor. Pacientul are de obicei o inhibiție a dezvoltării mentale și fizice. Boala este ereditară.
Din faptul că numele bolii are prefixul „pseudo”, puteți înțelege cu ușurință că imită hipoparatiroidismul. Al doilea nume al acestei boli este osteodistrofia ereditară a lui Albright, pe numele unui medic care a studiat și descris această patologie la mijlocul secolului trecut.
Tipuri de pseudohipoparatiroidism
Există două tipuri de pseudohipoparatiroidism - în funcție de dacă nivelul de calciu din sânge s-a modificat sau nu. Primul tip de boală Albright are același lucru Semne clinice, ca și în hipoparatiroidismul idiopatic, în timp ce o scădere a conținutului de calciu din sânge este caracteristică. Nu există sensibilitate tisulară la hormonul paratiroidian. Al doilea tip de pseudohipoparatiroidism, dimpotrivă, diferă prin faptul că nivelul de calciu este normal. Prin urmare, acest tip se numește pseudopseudohipoparatiroidism. Apropo, conform endocrinologilor, o formă poate trece cu ușurință în alta, în plus, membrii aceleiași familii pot avea tipuri diferite boli.
Cauzele pseudohipoparatiroidismului
Încălcări ale metabolismului fosfor-calciu se dezvoltă din cauza rezistenței țesuturilor la hormonul paratiroidian, care este produs de glandele paratiroide. Pe baza pseudohipoparatiroidismului a fost confirmat pentru prima dată fenomenul de afectare a sensibilității tisulare la un hormon produs de glandele endocrine sau care este introdus din exterior.
Pseudohipoparatiroidismul este o patologie de natură genetică. Este cauzată de un sindrom congenital - un receptor celular specific - hormon paratiroidian-adenilat ciclază, din cauza acestuia țesuturile periferice își pierd sensibilitatea la hormonul paratiroidian.
Cine este cel mai predispus la pseudohipoparatiroidism? În primul rând - copiii pacientului, alte rude, deoarece boala are o natură ereditară autosomal dominantă. Femeile suferă mai des de boala Albright. Experții explică acest lucru prin faptul că bărbații cu pseudohipoparatiroidism au fertilitate scăzută, iar ereditatea în sine trece de la tată la fiu doar în cazuri izolate.
Cât de des apare această boală - nu există statistici, aproximativ trei sute de cazuri sunt descrise în scrierile medicale.
Simptomele pseudohipoparatiroidismului
- Defecțiunile organelor sistemului endocrin cu pseudohipoparatiroidism provoacă, de asemenea, principalele semne: pacientul este de obicei îndesat, mai scund decât alții, din cauza membrelor inferioare scurtate, este obez, nivelul de glucoză din sânge este crescut, fața capătă o lună. -forma in forma;
- La reprezentanții sexului slab este încălcat ciclu menstrual. Adesea, aceste semne sunt însoțite de acromegalie, ginecomastie, diferite procese autoimune, sindromul Itsenko-Cushing și alte afecțiuni. Mai mare la astfel de pacienti si riscul de diabet si nu Diabet;
- Cei care suferă de boala Albright se plâng adesea de probleme cu dinții (au smalț deteriorat), pacienții sunt, de asemenea, îngrijorați de vărsături, convulsii tonice - atât spontane, cât și care apar sub influența iritanților, urina poate fi observată în sânge. Există riscul de cataractă. Caracterizat prin retard mintal. Copiii cu această boală nu fac față programului școlar, au memorie redusă;
- Modificări semnificative apar și în sistemul osteoarticular - apare osteoporoza difuză, se dezvoltă chisturi (tumori brune). Pacienții cu pseudohipoparatiroidism sunt mai predispuși să sufere de fracturi sau deformări osoase. Primul, al patrulea și al cincilea oase metacarpien și metatarsian sunt scurtate și se observă semne de resorbție osoasă pe mâini și picioare. În plus, calciul, care a fost eliberat din oase, începe să se acumuleze în țesutul subcutanat, formând calcificări (acest lucru se întâmplă și cu miozita osificantă), în zona articulațiilor, a cavității craniene. Calcificările sunt prezente și în mușchi, rinichi, miocard și pe pereții arterelor mari;
- Un alt semn distinctiv al bolii Albright este pigmentarea pielii. Pete maronii răspândite pe corp (atât pe o parte, cât și pe cealaltă parte).
În ceea ce privește capacitatea de a lucra, depinde de severitatea procesului și de eficacitate tratament medicamentos. Dacă forma bolii este ascunsă și nu există atacuri evidente, atunci capacitatea de a lucra este parțial păstrată. Dar există o serie de restricții - este imposibil, în special, să se lucreze cu mecanisme de mișcare, în transport, suprasolicitare neuropsihică, excesivă munca fizica. Dacă există atacuri convulsive frecvente, retardul mintal este clar exprimat, o deficiență vizuală semnificativă din cauza cataractei, atunci astfel de pacienți sunt deja considerați cu handicap.
Diagnosticul pseudohipoparatiroidismului
Simptomele sunt destul de specifice și dacă încep să apară, atunci trebuie să contactați imediat un medic calificat. Este devreme și diagnostic corect asigură succesul tratamentului.
Deoarece boala Albright este congenitală, este de obicei diagnosticată pacientului în copilărie (vârsta preșcolară, școlară primară). Geneticienii trag concluzii pe baza simptome clinice sunt folosite şi metode de diagnostic de laborator şi instrumental.
În special, pacientul donează sânge pentru a clarifica diagnosticul (la pacienții cu pseudohipoparatiroidism, nivelul de calciu este scăzut, iar fosforul și hormonul paratiroidian sunt mari, ridicate și cu activitate). fosfataza alcalinăîn sânge), urină (la un pacient, această analiză va arăta o scădere a fosforului și calciului excretați). De asemenea, se fac teste speciale care arată sensibilitatea pacientului a țesuturilor tubilor renali la hormonul paratiroidian. Uneori este nevoie să se evalueze nivelul de hidroxiprolină.
Metodele instrumentale implică diagnosticarea cu raze X (este cea mai informativă în identificarea modificărilor specifice ale țesuturilor osoase și musculare).
Tratamentul pseudohipoparatiroidismului
Această patologie ereditară rară este tratată cu suplimente de calciu. Doza este selectată de medic, în funcție de conținutul de calciu necesar pentru menținerea homeostaziei. Și pentru ca calciul să fie absorbit fără probleme, în regimul terapeutic sunt incluse și preparatele cu vitamina D. În plus, un medic pentru un pacient cu pseudohipoparatiroidism va dezvolta o dietă în care cantitatea de produse care conțin fluor va fi limitată. Această dietă va ajuta la normalizarea concentrației de calciu din sânge și va elimina semnele de hiperparatiroidism secundar.
Dacă boala lui Albright este însoțită de altele tulburări endocrine, atunci poate fi nevoie de terapie de substituție hormonală. Convulsiile sunt ameliorate cu o soluție de clorură de calciu sau gluconat de calciu, care se administrează intravenos. În cazul unei cataracte, un oculist este implicat în tratamentul acesteia, iar un psiholog ajută la eliminarea deficiențelor cognitive.
Cu o schemă de terapie bine gândită, prognosticul este foarte favorabil. Dar toți cei care au fost bolnavi de pseudohipoparatiroidism ar trebui să consulte în mod regulat geneticienii pentru a preveni boala la urmașii lor.
Prevenirea pseudohipoparatiroidismului
Avand in vedere ca boala este ereditara, singura modalitate de prevenire pentru cei care au avut pseudohipoparatiroidism in familie este vizitele regulate la medici geneticieni pentru consultatii.
- Tratamentul pseudohipoparatiroidismului
- Prevenirea pseudohipoparatiroidismului
- La ce medici ar trebui să vedeți dacă aveți pseudohipoparatiroidism?
Ce este pseudohipoparatiroidismul
Pseudohipoparatiroidism(Pseudēs greacă fals + hipoparatiroidism; sinonim: osteodistrofia ereditară Albright, boala Albright) - o boală ereditară rară a sistemului osos care imită hipoparatiroidismul și se caracterizează prin afectarea metabolismului calciului și fosforului; adesea însoțită de o întârziere a dezvoltării mentale și fizice.
Ce cauzează pseudohipoparatiroidismul?
Cauza pseudohipoparatiroidismului este un defect congenital - insensibilitatea țesuturilor periferice la acțiunea PTH.
Patogeneza (ce se întâmplă?) în timpul Pseudohipoparatiroidismului
Se crede că la baza pseudohipoparatiroidismului se află rezistența determinată genetic a rinichilor și a scheletului la acțiunea hormonului paratiroidian, ca urmare a unui defect în complexul unui anumit citoreceptor - hormon paratiroidian - adenilat ciclază, care perturbă formarea 3", 5"-AMP ciclic în rinichi, care este un mediator intracelular al acțiunii hormonului paratiroidian asupra proceselor metabolice. Pseudohipoparatiroidismul este o boală eterogenă genetic. La unii pacienți, citoreceptorul în sine este defect, care leagă hormonul paratiroidian (pseudohipoparatiroidism de tip Ia), la alții există un defect al proteinei de legare a nucleotidelor localizat în stratul dublu lipidic al membranei celulare și leagă funcțional receptorul de adenilat ciclază ( pseudohipoparatiroidism de tip Ib). Unii pacienți prezintă un deficit enzimatic al adenilat-ciclazei în sine (pseudohipoparatiroidism de tip II). Deficiența cAMP, rezultată din aceste defecte, duce la o încălcare a sintezei proteinelor specifice care determină efectul biologic al hormonului paratiroidian. Astfel, se pierde sensibilitatea organelor țintă, în special a rinichilor, la hormonul paratiroidian. Ca urmare, excreția urinară de fosfor scade, apare hiperfosfatemia, iar hipocalcemia se dezvoltă secundar. Deoarece glandele paratiroide sunt intacte în pseudohipoparatiroidism, ca răspuns la hipocalcemie, care stimulează producția de hormon paratiroidian, un secundar hiperparatiroidism. Formarea crescută a hormonului paratiroidian nu determină o creștere a excreției de fosfor și AMPc în urină din cauza rezistenței determinate genetic a tubilor renali la hormonul paratiroidian, dar este însoțită de modificări ale țesutului osos caracteristic hiperparatiroidismului, ceea ce indică menținerea sensibilității normale a osteoclastelor la hormonul paratiroidian. În cazul pseudohipoparatiroidismului, activitatea fosfatazei alcaline în serul sanguin este crescută sau este în intervalul normal (0,5-1,3). µmol fosfor anorganic per 1 ml ser de sânge pentru 1 h incubare la 37°; definiţie după Bodansky). Toate variantele de pseudohipoparatiroidism sunt o boală ereditară, natura moștenirii este autosomal dominantă. Fertilitatea scăzută a bărbaților care suferă de pseudohipoparatiroidism explică raritatea transmiterii acestuia de la tată la fiu; femeile sunt bolnave de 2 ori mai des decât bărbații.
De obicei, cu pseudohipoparatiroidism, se găsește hiperplazia compensatorie a glandelor paratiroide (prezența adenoamelor în acestea nu este tipică). În țesutul osos se observă modificări tipice hiperparatiroidismului - osteoporoză difuză, apariția chisturilor (așa-numitele tumori brune, tumori cu celule gigantice). Calciul eliberat din oase se depune sub formă de calcificări în țesutul subcutanat, precum și în rinichi, mușchi, miocard, pereții arterelor mari, conjunctiva ochiului și de-a lungul periferiei corneei.
Simptomele pseudohipoparatiroidismului
Semnele clinice ale pseudohipoparatiroidismului sunt similare cu cele ale idiopatiei hipoparatiroidism. Există atacuri de convulsii tonice care apar spontan sau sub influența oricăror stimuli. Calcificările subcutanate tind să se ulcereze. Osificarea subcutanată este adesea atât de pronunțată încât imită miozita osificantă. Caracterizat prin retard mintal, pipernicie, fața lunii, obezitate și brahidactilie, în special scurtarea primului, al patrulea și al cincilea metacarpien și a metatarsienilor. Pot exista exostoze multiple, dischondroplazie, manifestări de hiperparatiroidism secundar sub formă de resorbție subperiostală a oaselor degetelor; modificările epifizelor oaselor sunt aceleași ca în osteodisplazia fibroasă. Se observă adesea vărsături, precum și hematurie din cauza formării de pietre de oxalat în tractului urinar, dezvăluie cataractă lenticulară, hipoplazie a smalțului dentar.
La pacienții cu pseudohipoparatiroidism, împreună cu o scădere a sensibilității la hormonul paratiroidian a organelor țintă, rezistența la alți hormoni dependenți de sistemul adenilil ciclază, de exemplu, glandele sexuale la hormonii gonadotropi, glanda tiroidă la hormon de stimulare a tiroidei, organele țintă la glucagon și hormonul antidiuretic. Există o frecvență crescută boală autoimunăși diabetul zaharat observate hipotiroidism și hipertiroidism.
Există și pseudopseudohipoparatiroidismul, care se caracterizează prin absența hipocalcemiei, hiperfosfatemiei, convulsiilor și osteomalaciei.
Diagnosticul Pseudohipoparatiroidismului
Diagnosticul în cazurile tipice de boală se stabilește la copiii cu vârsta de 5-10 ani pe baza unui tablou clinic caracteristic, a anomaliilor multiple în dezvoltarea scheletului osos, a prezenței hipocalcemiei, a hiperfosfatemiei, a activității normale sau crescute a fosfatazei alcaline în serul sanguin, excreția redusă de calciu și fosfor în urină, conținut crescut de hormon paratiroidian în sânge. Prezența rezistenței tubulare renale la hormonul paratiroidian este confirmată de un test bazat pe determinarea cantității de fosfat și AMPc excretate în urină. Absența unei creșteri semnificative a conținutului de fosfați și AMPc în urină după administrarea hormonului paratiroidian la pacient indică rezistența rinichilor la acțiunea hormonului paratiroidian. La pacienţii cu hipoparatiroidism idiopatic şi postoperator, dimpotrivă, după administrare intravenoasă 200 de unități de hormon paratiroidian în urină, conținutul de fosfați și AMPc pentru 4 h creste de 2-10 ori fata de nivelul initial. Excreția urinară a hidroxiprolinei la pacienții netratați cu pseudohipoparatiroidism este normală sau ușor crescută, iar în cazul hipoparatiroidismului este redusă. Diagnosticul cu raze X al pseudohipoparatiroidismului se bazează pe detectarea modificărilor specifice ale oaselor și țesuturilor moi.
Pseudohipoparatiroidismul în combinație cu hipogonadismul la femei trebuie diferențiat de Sindromul Sereshevsky - Turner, cu care pseudopseudohipoparatiroidismul este similar fenotipic. Cu sindromul Sereshevsky-Turner, nu există cromatina sexuală, în locul ovarelor există fire de țesut conjunctiv care nu sunt detectate în timpul rectal și examenul cu ultrasunete.
Tratamentul pseudohipoparatiroidismului
Tratamentul hipocalcemiei constă în administrarea de suplimente de calciu în doze suficiente pentru a menține o concentrație normală de calciu în sânge. Terapia cu vitamina D este de mare importanță.Doza inițială este calculată de la 2000 UI / kg greutatea corporală pe zi, dar nu mai mult de 100.000 UI pe zi. Pentru a evita supradozajul preparatelor cu vitamina D, este necesar să se monitorizeze concentrația de calciu din sânge la fiecare 3-7 zile în primele două săptămâni de tratament și în fiecare lună în următoarele 2-3 luni. La atingerea unei concentrații stabile de calciu în sânge, este suficient să o verifici o dată la 2-3 luni. Puteți utiliza calcitrină, dihidrotachisterol, oxydevit, precum și alte medicamente ale formelor active de vitamina D. O dietă restricționată în fosfor ajută la normalizarea concentrației de calciu în sânge și la eliminarea simptomelor hiperparatiroidismului secundar. În caz de insuficiență a altor glande endocrine, se efectuează terapia de substituție cu hormoni corespunzători. Tratamentul cu hormon paratiroidian nu este eficient. Pentru ameliorarea crizelor convulsive se administrează intravenos o soluție 10% de clorură de calciu sau gluconat de calciu; în interior - 5-10% soluție de clorură de calciu, 1 lingură de 3-4 ori pe zi: gluconat de calciu, lactat de calciu - până la 10 Gîntr-o zi.
Prognosticul pentru terapia rațională este favorabil. Având în vedere natura ereditară a pseudohipoparatiroidismului, este necesară consilierea medicală genetică cu privire la posibilitatea apariției pseudohipoparatiroidismului la descendenți.
Pseudohipoparatiroidism
Ce este pseudohipoparatiroidismul?
Pseudohipoparatiroidism h(Pseudē greacă este fals + hipoparatiroidism; sinonim: osteodistrofia ereditară Albright, boala Albright) este o boală ereditară rară a sistemului osos care imită hipoparatiroidismul și se caracterizează prin afectarea metabolismului calciului și fosforului; adesea însoțită de o întârziere a dezvoltării mentale și fizice.
Ce provoacă / cauze ale pseudohipoparatiroidismului:
Cauza pseudohipoparatiroidismului este un defect congenital - insensibilitatea țesuturilor periferice la acțiunea PTH.
Patogeneza (ce se întâmplă?) în timpul pseudohipoparatiroidismului:
Se crede că la baza pseudohipoparatiroidismului se află rezistența determinată genetic a rinichilor și a scheletului la acțiunea hormonului paratiroidian, ca urmare a unui defect în complexul unui anumit citoreceptor - hormon paratiroidian - adenilat ciclază, care perturbă formarea 3", 5"-AMP ciclic în rinichi, care este un mediator intracelular al acțiunii hormonului paratiroidian asupra proceselor metabolice. Pseudohipoparatiroidismul este o boală eterogenă genetic. La unii pacienți, citoreceptorul în sine este defect, care leagă hormonul paratiroidian (pseudohipoparatiroidism de tip Ia), la alții există un defect al proteinei de legare a nucleotidelor localizat în stratul dublu lipidic al membranei celulare și leagă funcțional receptorul de adenilat ciclază ( pseudohipoparatiroidism de tip Ib). Unii pacienți prezintă un deficit enzimatic al adenilat-ciclazei în sine (pseudohipoparatiroidism de tip II). Deficiența cAMP, rezultată din aceste defecte, duce la o încălcare a sintezei proteinelor specifice care determină efectul biologic al hormonului paratiroidian. Astfel, se pierde sensibilitatea organelor țintă, în special a rinichilor, la hormonul paratiroidian. Ca urmare, excreția urinară de fosfor scade, apare hiperfosfatemia, iar hipocalcemia se dezvoltă secundar. Deoarece glandele paratiroide sunt intacte în pseudohipoparatiroidism, ca răspuns la hipocalcemie, care stimulează producția de hormon paratiroidian, un secundar hiperparatiroidism. Formarea crescută a hormonului paratiroidian nu determină o creștere a excreției de fosfor și AMPc în urină din cauza rezistenței determinate genetic a tubilor renali la hormonul paratiroidian, dar este însoțită de modificări ale țesutului osos caracteristic hiperparatiroidismului, ceea ce indică menținerea sensibilității normale a osteoclastelor la hormonul paratiroidian. În cazul pseudohipoparatiroidismului, activitatea fosfatazei alcaline în serul sanguin este crescută sau este în intervalul normal (0,5-1,3). µmol fosfor anorganic per 1 ml ser de sânge pentru 1 h incubare la 37°; definiţie după Bodansky). Toate variantele de pseudohipoparatiroidism sunt o boală ereditară, natura moștenirii este autosomal dominantă. Fertilitatea scăzută a bărbaților care suferă de pseudohipoparatiroidism explică raritatea transmiterii acestuia de la tată la fiu; femeile sunt bolnave de 2 ori mai des decât bărbații.
De obicei, cu pseudohipoparatiroidism, se găsește hiperplazia compensatorie a glandelor paratiroide (prezența adenoamelor în acestea nu este tipică). În țesutul osos se observă modificări tipice hiperparatiroidismului - osteoporoză difuză, apariția chisturilor (așa-numitele tumori brune, tumori cu celule gigantice). Calciul eliberat din oase se depune sub formă de calcificări în țesutul subcutanat, precum și în rinichi, mușchi, miocard, pereții arterelor mari, conjunctiva ochiului și de-a lungul periferiei corneei.
Simptomele pseudohipoparatiroidismului:
Semnele clinice ale pseudohipoparatiroidismului sunt similare cu cele ale idiopatiei hipoparatiroidism. Există atacuri de convulsii tonice care apar spontan sau sub influența oricăror stimuli. Calcificările subcutanate tind să se ulcereze. Osificarea subcutanată este adesea atât de pronunțată încât imită miozita osificantă. Caracterizat prin retard mintal, pipernicie, fața lunii, obezitate și brahidactilie, în special scurtarea primului, al patrulea și al cincilea metacarpien și a metatarsienilor. Pot exista exostoze multiple, dischondroplazie, manifestări de hiperparatiroidism secundar sub formă de resorbție subperiostală a oaselor degetelor; modificările epifizelor oaselor sunt aceleași ca în osteodisplazia fibroasă. Se observă adesea vărsături, precum și hematurie datorată formării de pietre de oxalat în tractul urinar, sunt detectate cataractă lenticulară și hipoplazie a smalțului dentar.
La pacienții cu spseudohipoparatiroidism, împreună cu o scădere a sensibilității la hormonul paratiroidian a organelor țintă, rezistența la alți hormoni dependenți de sistemul adenil-ciclazei, cum ar fi glandele sexuale la hormonii gonadotropi, glanda tiroidă la hormonul de stimulare a tiroidei, organele țintă la glucagon și hormon antidiuretic, pot fi observate. Există o frecvență crescută boală autoimunăși diabetul zaharat observate hipotiroidism și hipertiroidism.
Există și pseudopseudohipoparatiroidismul, care se caracterizează prin absența hipocalcemiei, hiperfosfatemiei, convulsiilor și osteomalaciei.
Diagnosticul pseudohipoparatiroidismului:
Diagnosticul în cazurile tipice de boală se stabilește la copiii cu vârsta de 5-10 ani pe baza unui tablou clinic caracteristic, a anomaliilor multiple în dezvoltarea scheletului osos, a prezenței hipocalcemiei, a hiperfosfatemiei, a activității normale sau crescute a fosfatazei alcaline în serul sanguin, excreția redusă de calciu și fosfor în urină, conținut crescut de hormon paratiroidian în sânge. Prezența rezistenței tubulare renale la hormonul paratiroidian este confirmată de un test bazat pe determinarea cantității de fosfat și AMPc excretate în urină. Absența unei creșteri semnificative a conținutului de fosfați și AMPc în urină după administrarea hormonului paratiroidian la pacient indică rezistența rinichilor la acțiunea hormonului paratiroidian. La pacienții cu hipoparatiroidism idiopatic și postoperator, dimpotrivă, după administrarea intravenoasă a 200 de unități de hormon paratiroidian în urină, conținutul de fosfați și AMPc timp de 4 h creste de 2-10 ori fata de nivelul initial. Excreția urinară a hidroxiprolinei la pacienții netratați cu pseudohipoparatiroidism este normală sau ușor crescută, iar în cazul hipoparatiroidismului este redusă. Diagnosticul cu raze X al pseudohipoparatiroidismului se bazează pe detectarea modificărilor specifice ale oaselor și țesuturilor moi.
Pseudohipoparatiroidismul în combinație cu hipogonadismul la femei trebuie diferențiat de Sindromul Shereshevsky-Turner, cu care pseudopseudohipoparatiroidismul este similar fenotipic. În sindromul Shereshevsky-Turner, nu există cromatina sexuală, în locul ovarelor există fire de țesut conjunctiv care nu sunt detectate prin examinarea rectală și cu ultrasunete.
Tratamentul pseudohipoparatiroidismului:
Tratamentul hipocalcemiei constă în administrarea de suplimente de calciu în doze suficiente pentru a menține o concentrație normală de calciu în sânge. Terapia cu vitamina D este de mare importanță.Doza inițială este calculată de la 2000 UI / kg greutatea corporală pe zi, dar nu mai mult de 100.000 UI pe zi. Pentru a evita supradozajul preparatelor cu vitamina D, este necesar să se monitorizeze concentrația de calciu din sânge la fiecare 3-7 zile în primele două săptămâni de tratament și în fiecare lună în următoarele 2-3 luni. La atingerea unei concentrații stabile de calciu în sânge, este suficient să o verifici o dată la 2-3 luni. Puteți utiliza calcitrină, dihidrotachisterol, oxydevit, precum și alte medicamente ale formelor active de vitamina D. O dietă restricționată în fosfor ajută la normalizarea concentrației de calciu în sânge și la eliminarea simptomelor hiperparatiroidismului secundar. În caz de insuficiență a altor glande endocrine, se efectuează terapia de substituție cu hormoni corespunzători. Tratamentul cu hormon paratiroidian nu este eficient. Pentru ameliorarea crizelor convulsive se administrează intravenos o soluție 10% de clorură de calciu sau gluconat de calciu; în interior - 5-10% soluție de clorură de calciu, 1 lingură de 3-4 ori pe zi: gluconat de calciu, lactat de calciu - până la 10 Gîntr-o zi.
Prognosticul pentru terapia rațională este favorabil. Având în vedere natura ereditară a pseudohipoparatiroidismului, este necesară consilierea medicală genetică cu privire la posibilitatea apariției pseudohipoparatiroidismului la descendenți.
Prevenirea pseudohipoparatiroidismului:
Ce medici ar trebui să contactați dacă aveți pseudohipoparatiroidism:
Ești îngrijorat de ceva? Doriți să aflați informații mai detaliate despre Pseudohipoparatiroidism, cauzele sale, simptomele, metodele de tratament și prevenire, cursul bolii și dieta după aceasta? Sau ai nevoie de o inspecție? Poti rezerva o programare la un medic– clinica Eurolaborator mereu la dispozitia ta! Cei mai buni doctori te examinează, studiază semne externeși ajută la identificarea bolii după simptome, vă sfătuiește și oferă avea nevoie de ajutor si pune un diagnostic. poti si tu sunați la un medic acasă. Clinica Eurolaborator deschis pentru tine non-stop.
Cum să contactați clinica:
Telefonul clinicii noastre din Kiev: (+38 044) 206-20-00 (multicanal). Secretarul clinicii va alege o zi și o oră convenabile pentru a vizita medicul. Coordonatele și direcțiile noastre sunt indicate. Uită-te mai detaliat despre toate serviciile clinicii pe ea.
(+38 044) 206-20-00
Dacă ați efectuat anterior vreo cercetare, asigurați-vă că duceți rezultatele la o consultație cu un medic. Dacă studiile nu au fost finalizate, vom face tot ce este necesar în clinica noastră sau cu colegii noștri din alte clinici.
Tu? Trebuie să fii foarte atent la sănătatea ta generală. Oamenii nu acordă suficientă atenție simptomele boliiși nu vă dați seama că aceste boli pot pune viața în pericol. Sunt multe boli care la început nu se manifestă în organismul nostru, dar în final se dovedește că, din păcate, este prea târziu să le tratăm. Fiecare boală are propriile simptome specifice, caracteristice manifestări externe- așa-zisul simptomele bolii. Identificarea simptomelor este primul pas în diagnosticarea bolilor în general. Pentru a face acest lucru, trebuie doar să faceți de mai multe ori pe an fi examinat de un medic nu numai pentru a preveni boală cumplită dar și pentru a menține o minte sănătoasă în corp și corpul în ansamblu.
Dacă vrei să pui o întrebare unui medic, folosește secțiunea de consultații online, poate că acolo vei găsi răspunsuri la întrebările tale și citește sfaturi de autoîngrijire. Dacă sunteți interesat de recenzii despre clinici și medici, încercați să găsiți informațiile de care aveți nevoie în secțiune. Înregistrați-vă și pe portalul medical Eurolaborator să fie în permanență la curent cele mai recente știriși actualizări ale informațiilor de pe site, care vă vor fi trimise automat prin poștă.
Alte boli din grupa Boli ale sistemului endocrin, tulburări de alimentație și tulburări metabolice:
| Criza Addisoniană (insuficiență suprarenală acută) |
| adenom mamar |
| Distrofia adipozogenitală (boala Perchkrantz-Babinski-Fröhlich) |
| Sindrom adrenogenital |
| Acromegalie |
| Nebunie alimentară (distrofie alimentară) |
| Alcaloza |
| Alcaptonurie |
| Amiloidoza (degenerarea amiloidului) |
| Amiloidoza stomacului |
| Amiloidoza intestinală |
| Amiloidoza insulelor pancreatice |
| Amiloidoza hepatică |
| Amiloidoza esofagiană |
| Acidoza |
| Malnutriție proteico-energetică |
| Boala celulelor I (mucolipidoză tip II) |
| Boala Wilson-Konovalov (distrofie hepatocerebrală) |
| Boala Gaucher (lipidoza glucocerebrozidica, glucocerebrozidoza) |
| boala Itsenko-Cushing |
| boala Krabbe (leucodistrofie cu celule globoide) |
| boala Niemann-Pick (sfingomielinoză) |
| boala Fabry |
| Gangliozidoza GM1 tip I |
| Gangliozidoza GM1 tip II |
| Gangliozidoza GM1 tip III |
| Gangliozidoza GM2 |
| Gangliozidoza GM2 tip I (idioție amaurotică Tay-Sachs, boala Tay-Sachs) |
| Gangliozidoza GM2 tip II (boala Sandhoff, idiotia amaurotica Sandhoff) |
| Gangliozidoza GM2 juvenila |
| Gigantism |
| Hiperaldosteronism |
| Hiperaldosteronism secundar |
| Hiperaldosteronism primar (sindromul Conn) |
| Hipervitaminoza D |
| Hipervitaminoza A |
| Hipervitaminoza E |
| Hipervolemie |
| Comă hiperglicemică (diabetică). |
| Hiperkaliemie |
| Hipercalcemie |
| Hiperlipoproteinemie de tip I |
| Hiperlipoproteinemie tip II |
| Hiperlipoproteinemie tip III |
| Hiperlipoproteinemie de tip IV |
| Hiperlipoproteinemie de tip V |
| Comă hiperosmolară |
| Hiperparatiroidism secundar |
| Hiperparatiroidismul primar |
| Hiperplazia timusului (glanda timus) |
| Hiperprolactinemie |
| hiperfuncție testiculară |
| Hipercolesterolemie |
| hipovolemie |
| Comă hipoglicemică |
| hipogonadism |
| Hipogonadism hiperprolactinemic |
| Hipogonadism izolat (idiopatic) |
| Hipogonadism primar congenital (anorhism) |
| Hipogonadism, primar dobândit |
| hipokaliemie |
| Hipoparatiroidismul |
| hipopituitarism |
| Hipotiroidismul |
| Glicogenoză tip 0 (aglicogenoză) |
| Glicogenoza tip I (boala lui Girke) |
| Glicogenoza tip II (boala Pompe) |
| Glicogenoză tip III (boala rujeolă, boala Forbes, dextrinoză limită) |
| Glicogenoză tip IV (boala Andersen, amilopectinoză, glicogenoză difuză cu ciroză hepatică) |
| Glicogenoza tip IX (boala lui Hag) |
| Glicogenoza de tip V (boala McArdle, deficit de miofosforilază) |
| Glicogenoza de tip VI (boala Hers, deficit de hepatofosforilază) |
| Glicogenoza de tip VII (boala Tarui, deficit de miofosfofructokinaza) |
| Glicogenoza tip VIII (boala lui Thomson) |
| Glicogenoza tip XI |
| Glicogenoza de tip X |
| Deficiență (insuficiență) de vanadiu |
| Deficiență (insuficiență) de magneziu |
| Deficiență (insuficiență) de mangan |
| Deficiență (insuficiență) de cupru |
| Deficiență (insuficiență) de molibden |
| Deficiență (insuficiență) de crom |
| deficiență de fier |
| Deficit de calciu (deficit alimentar de calciu) |
| Deficit de zinc (deficit alimentar de zinc) |
| comă cetoacidotică diabetică |
| Disfuncția ovariană |
| Gușă difuză (endemică). |
| Pubertate întârziată |
| Excesul de estrogen |
| Involuția glandelor mamare |
| Nanism (statură mică) |
| Kwashiorkor |
| Mastopatie chistică |
| xantinurie |
| Comă lactică |
| Leucinoza (boala siropului de artar) |
| Lipidoze |
| lipogranulomatoza Farber |
| Lipodistrofie (degenerare grasă) |
| Lipodistrofie congenitală generalizată (sindrom Sape-Lawrence) |
| Lipodistrofie hipermusculară |
| Lipodistrofie după injecție |
| Lipodistrofie progresivă segmentară |
| Lipomatoza |
| Lipomatoza dureroasa |
| Leucodistrofie metacromatică |
| Comă mixedem |
