E.V. Tozliyan, detský endokrinológ, genetik, PhD, I.V. Shulyakova, neurologička, PhD,
izolovaný štrukturálne členenie"Vedecko-výskumný klinický ústav pediatrie" SBEE HPE "Ruská národná výskumná lekárska univerzita pomenovaná po N.I. Pirogov“ z Ministerstva zdravotníctva Ruskej federácie, Moskva
Kľúčové slová:
deti, pseudohypoparatyreóza, Albrightova dedičná osteodystrofia, obezita, hypokalciémia, diagnostika, rezistencia na parathormón.
Kľúčové slová: deti, pseudohypoparatyreóza, Albrightova dedičná osteodystrofia, obezita, hypokalciémia, diagnostika, rezistencia na parathormón.
Pseudohypoparatyreóza (grécky pseudes – nepravda + hypoparatyreóza; synonymum: Albrightova dedičná osteodystrofia, syndróm „jávskeho kurčaťa“) – zriedkavé dedičné ochorenie kostrový systém, ktorý napodobňuje hypoparatyreózu a je charakterizovaný poruchou metabolizmu vápnika a fosforu; často sprevádzané mentálnou retardáciou a fyzický vývoj. Toto ochorenie prvýkrát opísal americký endokrinológ Albright F. v roku 1942. Prevalencia ochorenia je 7,9 na 1 milión ľudí.
GENETICKÉ ÚDAJE
Pseudohypoparatyreóza (PHP) je geneticky heterogénne ochorenie. Údaje o type dedičného prenosu sú protichodné: dominantné aj autozomálne dominantné, autozomálne recesívne typy. Vo väčšine prípadov je rozvoj Albrightovej dedičnej osteodystrofie spojený s mutáciami v lokuse 20q13 génu GNAS1 lokalizovaného na chromozóme 20 (Patten et al., 1990), ktorý kóduje Gs-alfa proteín asociovaný s parathormónom (PTH) receptor. Podobný fenotyp bol zistený aj u pacientov s intersticiálnou deléciou dlhého ramena chromozómu 2 lokusu 2q37.
PATOGENÉZA
Patogenéza pseudohypoparatyreózy je založená na geneticky podmienenej odolnosti obličiek a skeletu voči pôsobeniu parathormónu v dôsledku defektu komplexu „špecifický cytoreceptor – parathormón – adenylátcykláza“, ktorý narúša tvorbu cyklických 3“- 5"-adenozínmonofosfát (cAMP) v obličkách, ktorý je intracelulárnym mediátorom účinku parathormónu na metabolické procesy. Pseudohypoparatyreóza je geneticky podmienená heterogénne ochorenie. U niektorých pacientov je defektný samotný cytoreceptor, ktorý viaže parathormón (pseudohypoparatyreóza typu 1A), u iných je defekt proteínu viažuceho nukleotidy lokalizovaného v lipidovej dvojvrstve bunkovej membrány a funkčnej väzby receptora na adenylátcyklázu ( pseudohypoparatyreóza typu 1B). Niektorí pacienti majú enzymatický deficit samotnej adenylátcyklázy (pseudohypoparatyreóza typu 2). Nedostatok cAMP, ktorý je výsledkom týchto defektov, vedie k narušeniu syntézy špecifických proteínov, ktoré určujú biologický účinok parathormónu. Tým sa stráca citlivosť cieľových orgánov na parathormón.
KLINICKÉ CHARAKTERISTIKY
V súčasnosti sú 4 klinické formy patológie: typy 1A, 1B, 1C a 2. Znalosť ich klinických a biochemických znakov a údajov genetický výskum umožňuje diferenciálnu diagnostiku v rámci samotnej nozologickej formy.
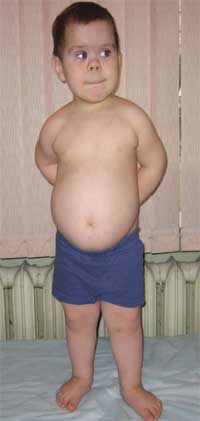
Bežné znaky, ktoré umožňujú podozrenie na ochorenie, sú neprimeraný fyzický vývoj, nízky vzrast (až trpaslík) v dôsledku skrátenia dolných končatín(foto 1), brachydaktýlia (foto 2), okrúhla tvár v tvare mesiaca (foto 3). Niekedy sa vyskytujú exostózy a aplázia zubov.
Fotografia 1.
Vzhľad dieťa s Albrightovou osteodystrofiou
(vlastnosti fenotypu, nízky vzrast v dôsledku skrátenia dolných končatín)
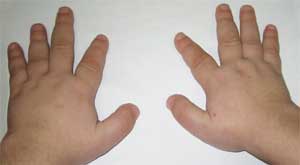
Fotografia 2.
Vlastnosti kostrového systému u pacienta
s Albrightovou osteodystrofiou
(brachydaktýlia - krátke prsty)
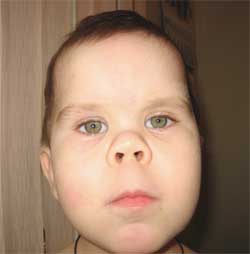
Fotografia 3.
Vlastnosti fenotypu dieťaťa
s Albrightovou osteodystrofiou
(okrúhla "mesiaca" tvár)
Prudké skrátenie I, III a V záprstných a metatarzálnych kostí (najmä III a IV) sa považuje za patognomický znak, v dôsledku čoho sú prsty II na rukách a nohách dlhšie ako ostatné a keď je ruka zovreté v päsť, v oblasti IV a V metakarpofalangeálnych kĺbov nie sú žiadne vydutiny - takzvaný brachymetafalangizmus. Zisťujú sa aj krátke široké falangy, zhrubnutie lebečnej klenby a demineralizácia kostí (osteoporóza), obezita.
Mentálna retardácia (často stredne závažná) sa nachádza približne u 20 % pacientov. Podľa niektorých autorov sa oligofrénia vyskytuje v 70% prípadov s hypokalciémiou a v 30% prípadov s normokalciémiou. mentálne procesy pacienti sú pomalí. V neurologickom stave sú často zaznamenané motorická nemotornosť, neurotické reakcie: strach, úzkosť, nepokoj, zlý sen, zvýšené reflexy, kŕče, ktoré majú tetanický charakter a sú spôsobené hypokalciémiou, niekedy kŕčovité záchvaty. Popisujú sa aj myopatické príznaky: svalová únava, svalová slabosť. Často sa pozorujú extrapyramídové poruchy: choreiformná hyperkinéza, atetóza, tvárový hemispazmus, parkinsonizmus, v niektorých prípadoch sa vyskytujú epileptické paroxyzmy, cerebelárne príznaky: ataxia, zhoršená koordinácia.
Často sa stanovujú kalcifikácie mäkkých tkanív, podkožné kalcifikácie (hrudník, brucho, šľachy kalkanea), ktorých histologické vyšetrenie - osteóm kože(Izraeli et al., 1992), mozog (bazálne gangliá). Je dôležité poznamenať, že kalcifikácie môžu byť prítomné už pri narodení. V dôsledku hypokalcémie zvyčajne vzniká šedý zákal a vznikajú defekty zubnej skloviny.
PSEUDOHYPOPARATYROIZA TYPU 1A
má autozomálne dominantný spôsob dedičnosti. Gén pseudohypoparatyreoidizmu typu 1A, GNAS1, sa nachádza na dlhom ramene chromozómu 20, v lokuse 20q13.2. Rozvoj ochorenia je spojený s nedostatkom proteínu viažuceho guanín-nukleotid (Gs-proteín). Súčasne PTH, viažuci sa na receptory cieľových tkanív, nie je schopný aktivovať cyklický adenozínmonofosfát (cAMP) a spôsobiť tkanivovú odpoveď. Pravdepodobne podobný mechanizmus je základom rozvoja necitlivosti tkanív iných orgánov a žliaz s vnútornou sekréciou (hypofunkcia štítna žľaza pohlavné žľazy, hypofýza, diabetes mellitus, ako aj znížená odpoveď pečene na podanie glukagónu), pozorované pri pseudohypoparatyreóze typu 1A. Pri tomto type patológie nedochádza k zvýšenému vylučovaniu cAMP močom, čo je charakteristické pre normu, ako odpoveď na exogénne podávanie PTH. Ochorenie je diagnostikované častejšie vo veku 5-10 rokov. Pacienti majú nízky vzrast, krátky krk, okrúhlu tvár, skrátenie záprstných a metatarzálnych kostí (častejšie skrátenie IV a menej často II prstov) - tzv. brachy-metafalangizmus. Zaznamenáva sa kalcifikácia mäkkých tkanív, podkožné kalcifikácie, ktoré možno zistiť už pri narodení; často dochádza k súčasnému postihnutiu ďalších žliaz s vnútornou sekréciou: štítnej žľazy (hypofunkcia), pohlavných žliaz, pankreasu (diabetes mellitus). V dôsledku hypokalcémie často vzniká šedý zákal a defekt zubnej skloviny. Ako diferenciálny diagnostický test na rozdiel medzi PGP typu 1A a hypoparatyreózou: absencia klinického účinku parenterálneho podávania PTH vo forme zvýšenia hladiny vápnika v krvi a zvýšenej renálnej exkrécie fosforu v moč (fosfaturický účinok).
Biochemickým vyšetrením sa zistí hypokalciémia, hyperfosfatémia, zvýšenie hladiny parathormónu v krvi a hypofosfatúria. Hladina Gs-proteínu v krvi je znížená. o röntgenové vyšetrenie kostrového systému sa zisťuje skrátenie záprstných a metatarzálnych kostí, generalizovaná demineralizácia, zhrubnutie kostí lebečnej klenby.
PSEUDOHYPOPARATYROIZA TYPU 1B
má autozomálne dominantný typ dedičnosti, nie je však vylúčený dominantný, X-viazaný typ dedičnosti. Je potrebné mať na pamäti niekedy pozorovanú neúplnú penetráciu génu choroby a možnosť skrytého nosiča patológie. Preto sa odporúča klinické (zistenie subklinického priebehu ochorenia) a biochemické vyšetrenie (stanovenie hladín vápnika, fosforu, PTH v krvi) podozrivých nosičov ochorenia. HPT typu 1B je spôsobený nedostatkom tkanivových receptorov pre parathormón v cieľových orgánoch a obmedzenou rezistenciou na parathormón. Klinický obraz je podobný ako na klinike typu 1A, nedochádza však k poškodeniu iných žliaz s vnútornou sekréciou, menej častá je osteodystrofia.
U pacientov nedochádza k reakcii obličiek na exogénne podanie parathormónu v podobe zvýšeného vylučovania cyklického adenozínmonofosfátu močom, avšak na rozdiel od typu 1A je hladina Gs-proteínu v krvi v norme. Ženy sú postihnuté častejšie ako muži, ale závažnosť ochorenia môže byť rovnaká u mužov aj žien.
PSEUDOHYPOPARATYROIZA TYPU 1C
niektorí autori sa stotožňujú s pseudo-pseudohypoparatyreoidizmom (PPHP), ktorý opísal Albright F. v roku 1952. Vyznačuje sa klinickým obrazom charakteristickým pre PAH, ale hladiny vápnika, fosforu v krvi a moči zostávajú v rámci normy. Ukazovatele PTH a Gs-proteínu v krvi sú tiež uložené na normálna úroveň. Niektorí pacienti s PHP typu 1C majú delécie de novo na chromozóme 2. Je možné, že tento variant ochorenia je podtypom HPP typu 1A.
PSEUDOHYPOPARATYROIZA TYPU 2
klinicky podobné iným typom ochorenia, ale má autozomálne recesívny spôsob dedičnosti. Existencia autozomálne dominantných foriem patológie nie je vylúčená. Patogenéza vývoja je spojená s vnútrobunkovou rezistenciou na cAMP. V tomto prípade sa PTH viaže na receptory a spôsobuje normálnu bunkovú odpoveď na PTH vo forme zvýšenia vylučovania cAMP. Intracelulárna necitlivosť na cAMP však neumožňuje plnú realizáciu účinku PTH. Zároveň je zachovaná normálna reakcia obličiek na exogénne podanie parathormónu vo forme zvýšeného vylučovania cyklického adenozínmonofosfátu močom. Bolo navrhnuté, že HWP typu 2 môže súvisieť s nedostatkom vitamínu D.
Identifikované typy PHP sa teda klinicky vyznačujú zníženou citlivosťou cieľových orgánov na PTH, líšia sa však patogenetickými mechanizmami vzniku tkanivovej necitlivosti.
DIAGNOSTIKA
Povaha renálnej exkrécie cAMP v reakcii na podanie PTH môže slúžiť ako laboratórne diferenciálne diagnostické vyšetrenie: zvýšené vylučovanie cAMP je zaznamenané u typu 2 a jeho absencia u typu 1. Diagnóza je potvrdená detekciou zníženej hladiny guanín-nukleotidu -väzbový proteín (Gs-proteín) v krvi (v priemere 1,5-2 krát) v porovnaní s normou. Hypokalciémia je zvyčajne spojená s hyperfosfatémiou a hypofosfatúriou. Hladina PTH je zvýšená; pri type 1C je hladina PTH v norme, z čoho vznikol názov „pseudohypoparatyreóza“. Röntgenovým vyšetrením kostrového systému sa zistí skrátenie záprstných a metatarzálnych kostí, často generalizovaná demineralizácia (osteoporóza), zhrubnutie kostí lebečnej klenby. Dermatoglyfická kresba ukazuje posun axiálneho palmárneho triradia.
Kritériá diagnózy:
- nízky rast;
- okrúhla tvár;
- oneskorenie nervozity duševný vývoj;
- kostrové anomálie;
- nízka hladina vápnika v sére;
- vysoká hladina parathormónu v krvi;
- znížené vylučovanie fosfátu a cAMP močom.
LIEČBA A PREVENCIA
Liečba hypokalciémie spočíva v vymenovaní doplnkov vápnika v dávkach dostatočných na udržanie normálnej koncentrácie vápnika v krvi. Veľký význam má terapiu vitamínom D. V súčasnosti sa používajú aktívne metabolity vitamínu D - oksidevit, 1-alfa-D3, kalcitrín atď.v dávke 1-2 mcg/deň s pozitívnym výsledkom (zvýšenie vápnika v krvi, zníženie prejavov tzv. konvulzívny syndróm). Účinný je aj tachystín (0,5–1,5 mg/deň). Tento liek zvyšuje vstrebávanie vápnika v črevách a tým zvyšuje hladinu vápnika v krvi. Antikonvulzívna terapia sa používa ako dodatočná liečba. Liečba nemá výrazný vplyv na intelektuálny vývoj, ale spolu s poklesom symptómov konvulzívneho syndrómu sa pozoruje regresia neurologických prejavov (subkortikálne poruchy, choreiformná hyperkinéza, atetóza atď.). Aby sa predišlo predávkovaniu prípravkami vitamínu D, je potrebné kontrolovať koncentráciu vápnika v krvi každé 3-7 dní počas prvých 2 týždňov liečby a každý mesiac počas nasledujúcich 2-3 mesiacov. Po dosiahnutí stabilnej koncentrácie vápnika v krvi ho stačí kontrolovať raz za 2-3 mesiace. Diéta s obmedzeným obsahom fosforu pomáha normalizovať koncentráciu fosforu aj vápnika v krvi a eliminovať príznaky sekundárnej hyperparatyreózy. V prípade nedostatočnosti iných žliaz s vnútornou sekréciou, substitučná liečba zodpovedajúce hormóny.
Liečba parathormónom je neúčinná. Na zmiernenie konvulzívnych záchvatov sa intravenózne podáva 10% roztok chloridu vápenatého alebo glukonátu vápenatého; vnútri - 5-10% roztok chloridu vápenatého, 1 polievková lyžica 3-4 krát denne: glukonát vápenatý, laktát vápenatý - do 10 g denne.
PREDPOVEĎ pre život je určená závažnosťou konvulzívneho syndrómu.
PREVENCIA ochorenie je založené na údajoch lekárskeho genetického poradenstva.
LEKÁRSKE GENETICKÉ PORADENSTVO
V medicínskom genetickom poradenstve treba vychádzať z autozomálne dominantného typu dedičnosti a vysokého (50%) rizika recidívy ochorenia v rodine s dedičnými formami. Aby bolo možné identifikovať povahu typu dedičnosti, je potrebné vykonať dôkladné vyšetrenie rodičov, pretože syndróm sa môže prejaviť s minimálnymi klinickými príznakmi. V súčasnosti je vyvinutá a zdokonaľovaná molekulárno-genetická diagnostika ochorenia typizáciou mutácií v géne GNAS1 na chromozóme 20. Vyvíjajú sa metódy prenatálnej diagnostiky ochorenia ako celku a jeho jednotlivých typov.
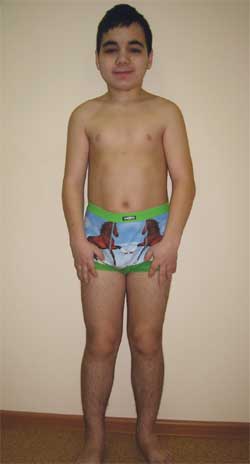
KLINICKÉ POZOROVANIE Chlapec G., 14,5 roka (foto 4), bol prijatý do Výskumného klinického ústavu detských chorôb s diagnózou degeneratívne ochorenie. nervový systém? vrodený vonkajší hydrocefalus; symptomatická epilepsia; dedičný syndróm? skladovacia choroba? metabolická encefalopatia; subklinická hypotyreóza; nízky vzrast zmiešanej genézy; kognitívne poruchy.
Sťažnosti pri prijatí k intenzívnym záchvatovým bolestiam hlavy lokalizovaným v čelná oblasť a sprevádzané zvracaním, ktoré prináša úľavu, znížená pamäť a školský výkon, kŕčovité záchvaty, pri ktorých dochádza k zášklbom v pravej ruke.
Fotografia 4.
Dieťa G., 14,5 roka, s Albrightovou osteodystrofiou
(znaky fenotypu, nízky vzrast, skrátenie končatín, brachydaktýlia)
Rodinná história: rodičia sú Arméni podľa národnosti, nie sú pokrvne príbuzní a nemajú žiadne profesionálne riziká. V rodokmeni prípadov duševná choroba, epilepsia, vývojové oneskorenia neboli zaznamenané. Sibs, sestra, 17 rokov - podľa slov - zdravá.
História života a choroby: chlapček z 2.tehotenstva,ktoré prebiehalo bez čŕt,druhý pôrod,načas,fyziologický,pôrodná váha -3100g,dĺžka -51cm.Hneď zakričal,Apgar skóre -7/9 bodov. Zhoršenie na 3. deň - novorodenecké kŕče, zastavené v pôrodnici. Skoré postnatálne obdobie - bez rysov. V prvom roku života došlo k miernemu oneskoreniu tempa vo vývoji motoriky, samostatná chôdza od 1 roka 3 mesiacov. V tejto súvislosti bol sledovaný neurológom s diagnózou organického poškodenia centrálneho nervového systému; vrodený hydrocefalus; neonatálne kŕče; febrilné kŕče v histórii.
Prijatý diakarb, finlepsin. Debut záchvatov od 1 roka 11 mesiacov. – asymetrické, tonikum vo forme napätia pravá ruka a nohy, so vznikom očí, do 2 minút, bez straty vedomia, často do 10 epizód denne. Depakine dostával nepravidelne. Na pozadí sebazrušenia - jediný tonický stav. V 2 rokoch bolo v mieste bydliska urobené CT mozgu, kde boli zistené jednotlivé ložiská demyelinizácie v okcipitálnych lalokoch.
Konzultované s neurochirurgom, odporúčaná konzervatívna liečba. Od 3 rokov dochádza k oneskoreniu psychoverbálneho vývinu, odporúča sa dohľad psychiatra.
Vo veku 4–5 rokov si rodičia začali všímať deformáciu a skrátenie prstov na nohách a rukách, najmä II–IV prstov symetricky na rukách a nohách, a zníženie rýchlosti rastu. Vo veku 8 rokov záver logopéda - všeobecné porušenie rečový prejav 2.-3.stupňa, školenie v špecializovanej škole. V rovnakom veku vyšetrenie genetikom v mieste bydliska, záver: dedičné ochorenie výmena? odporučila sa štúdia aminokyselín v krvi, nezistili sa žiadne zmeny; konečný záver: údaje o dedičnom metabolickom ochorení neboli odhalené; hypochondroplázia; odporúčaná liečba neurológom a endokrinológom.
Tabuľka.
Profil duševného vývoja dieťaťa G., 14,5 ročného (IQ = 68)
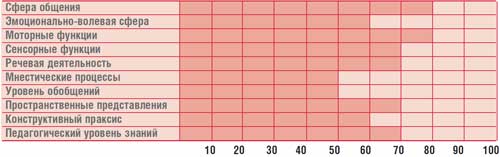
Vo veku 8 rokov bol u endokrinológa konzultovaný o oneskorení rastu a vývoja. Pri röntgenovom vyšetrení rúk boli zaznamenané nasledujúce znaky: stredné, hlavné falangy a metakarpálne kosti boli skrátené a zhrubnuté; Diagnóza rádiológa znela achondroplázia.
Opakovane vyšetrený v mieste bydliska v neurologickej nemocnici. V 12 rokoch sa objavili kŕčové záchvaty bez straty vedomia s trhaním pravej ruky, ktoré sú sériového charakteru, bola predpísaná antikonvulzívna terapia (Depakine), frekvencia záchvatov sa výrazne znížila. V 13 rokoch bola vykonaná MRI mozgu s kontrastom - symetrické zmeny na báze spánkových lalokov na úrovni jadier v podobe zvýšenia signálu MR, ktorý je typický pre toxické (mangán) alebo metabolické (meď, železo) encefalopatie.
Opakované vyšetrenie vo veku 13 rokov 3 mesiace. endokrinológ, pri štúdiu profilu štítnej žľazy bolo zistené zvýšenie hormónu stimulujúceho štítnu žľazu (TSH), diagnostikovaná subklinická hypotyreóza a predpísaný L-tyroxín.
Pri analýze ambulantnej karty a dokumentácie dieťaťa v mieste bydliska bola štúdia vápnika a fosforu vykonaná raz, vo veku 1,5 roka bola zaznamenaná hypokalcémia, ale túto príležitosť následné vyšetrenie nebolo vykonané. Vzhľadom na neistotu diagnózy v mieste bydliska poslal genetik dieťa do Moskvy, do Vedeckého výskumného klinického ústavu pediatrie, aby objasnil diagnózu.
Údaje z objektívneho výskumu:
Výška - 143 cm, hmotnosť - 43 kg.
Telesný vývoj je veľmi nízky, harmonický, telesná stavba je vzhľadom na skrátenie končatín neprimeraná. Rast Sds zodpovedá -2,8 odchýlkam od normy (normálne -2 + 2).
Fenotypové znaky: okrúhla tvár, krátky krk, antimongoloidný rez palpebrálnych štrbín, široký nosový mostík, vysoké čelo, brachydaktýlia, skrátenie IV a V metakarpálnych a metatarzálnych kostí (foto 5). Na vnútorných orgánoch - bez funkcií. Sexuálny vývoj - Tanner III-IV štádium (čo zodpovedá veku).
Údaje z laboratórnych a funkčných štúdií:
Klinická analýza krv a moč sú normálne.
Biochemický krvný test: celkový vápnik - 1,39 (normálne 2,02-2,6 mmol / l), ionizovaný vápnik - 0,61 (normálny 1,13-1,32 mmol / l), anorganický fosfor - 3,66 (norma 0,86-1,56 mmol / l), ostatné ukazovatele sú v rámci normálny rozsah.
Biochemický rozbor moču: vylučovanie fosfátov obličkami je znížené - 11,5 mmol/l (norma 19-32 mmol/l).
Profil štítnej žľazy: TSH - 11,75 (norma 0,4-4,0 μIU / ml), voľný T4 - 0,49 (norma 1,0-1,8 ng / dl).
Parathormón - 499 (normálne 12-65 pg / ml), rastový hormón - 7 ng / ml (normálne 7-10 ng / ml), somatomedín-C - 250 ng / ml (normálne 88-360 ng / ml).
ultrazvuk vnútorné orgány- bez funkcií.
EKG - migrácia supraventrikulárneho kardiostimulátora na pozadí pravidelnej srdcovej frekvencie 71–80 úderov/min. Neúplná blokáda pravej nohy jeho zväzku. Porušenie procesu repolarizácie v myokarde zadná stenaľavej komory (znížená z.T III, aVF).
R-grafia chrbtice - pravostranná skolióza hrudný chrbtica 1. stupňa, ťažká osteoporóza.
R-grafia rúk so zachytením predlaktí - skrátenie a rozšírenie koncových a stredných falangov. Kostný vek - 13,5-14 rokov.
EEG vzory epileptickej aktivity neboli zaregistrované.
MRI mozgu - MRI obraz mnohopočetných subkortikálnych ložísk zvýšeného MR signálu v predné laloky, vonkajší kompenzovaný hydrocefalus s atrofiou mozgovej substancie.
MSCT mozgu - symetrické oblasti kalcifikácie lentiformných jadier. Difúzne hyperdenzné oblasti v talame, kaudátové jadrá s oblasťou kalcifikácie vpravo. Viacnásobné bodkované kalcifikácie vnútorných mäkkých tkanív lebky.
Audiogram - bez patológie.
Prebieha diagnostika DNA v géne GNAS1.
Odborná rada:
Endokrinológ - Albrightova dedičná osteodystrofia typu 1A (pseudohypoparatyreóza). Primárna hypotyreóza, neúplné lekárske odškodnenie.
Oftalmológ - úplná sekundárna katarakta. Odporúčané chirurgická liečba.
Psychológ – kognitívne poruchy (psychologický profil dieťaťa je uvedený v tabuľke).
Berúc do úvahy fenotyp dieťaťa, údaje o anamnéze, výsledky dodatočný výskum(hypokalcémia, hyperfosfatémia, hypofosfatúria, zvýšená hladina parathormónu v krvi), kalcifikácie v substancii mozgu, prítomnosť katarakty, hypotyreóza), bola stanovená diagnóza: Albrightova dedičná osteodystrofia typu 1A (pseudohypoparatyreóza). Odporúča sa vykonať DNA diagnostiku – hľadanie mutácií v géne GNAS1.
Liečba: dieťaťu sa odporúča užívať euthyrox v dávke 100 mcg / deň; aktívny metabolit vitamínu D - alfa-D3 ("Teva") v dávke 2 mcg / deň; vápnik ("Sandoz") 2000 mg / deň; konštantný príjem antikonvulzívnej terapie - finlepsin 800 mg / deň pod dohľadom neurológa-epiptológa; triedy s logopedológom-defektológom a psychológom; energotropná terapia (Elkar a koenzým Q10 vo vekových dávkach). Kontrola ukazovateľov metabolizmu fosforu a vápnika, hladiny parathormónu.
Touto cestou, Prezentované klinické pozorovanie demonštruje zložitosť diferenciálneho diagnostického vyhľadávania, dôležitosť včasného štúdia jednoduchých biochemických parametrov (pri epilepsii je povinný opakovaný skríning ukazovateľov metabolizmu fosforu a vápnika), výstupy neskorej diagnostiky geneticky podmieneného ochorenia. , potreba integrácie jednotlivých znakov do celkového fenotypu konkrétneho patologického stavu pre cielenú včasnú diagnostiku určitých foriem dedičných ochorení. Včasná diagnostika, objasnenie genézy každého syndrómu sú obzvlášť dôležité, pretože umožňujú nájsť najlepší prístup k liečbe týchto stavov, prevencii možné komplikácie(až do zdravotného postihnutia dieťaťa); prevencia recidívy dedičných chorôb v postihnutých rodinách (lekárske genetické poradenstvo). To diktuje potrebu lekárov rôznych špecialít, aby jasne navigovali tok dedičnej patológie. Bibliografia je v štádiu revízie.
Pseudohypoparatyreóza - dosť zriedkavé ochorenie kostrový systém, ktorého podstatou je porušenie výmeny vápnika a fosforu. Pacient má zvyčajne inhibíciu duševného a fyzického vývoja. Choroba je dedičná.
Z toho, že názov choroby má predponu „pseudo“, ľahko pochopíte, že napodobňuje hypoparatyreózu. Druhým názvom tejto choroby je Albrightova dedičná osteodystrofia, podľa mena lekára, ktorý študoval a opísal túto patológiu v polovici minulého storočia.
Typy pseudohypoparatyreózy
Existujú dva typy pseudohypoparatyreózy – v závislosti od toho, či sa hladina vápnika v krvi zmenila alebo nie. Prvý typ Albrightovej choroby má to isté Klinické príznaky, ako pri idiopatickej hypoparatyreóze, pričom charakteristický je pokles obsahu vápnika v krvi. Neexistuje žiadna citlivosť tkaniva na parathormón. Druhý typ pseudohypoparatyreózy sa naopak líši v tom, že hladina vápnika je normálna. Preto sa tento typ nazýva pseudopseudohypoparatyreóza. Mimochodom, jedna forma môže podľa endokrinológov ľahko prejsť do druhej, navyše členovia tej istej rodiny môžu mať odlišné typy choroby.
Príčiny pseudohypoparatyreózy
Porušenie metabolizmu fosforu a vápnika sa vyvíja v dôsledku rezistencie tkaniva na parathormón, ktorý produkujú prištítne telieska. Na podklade pseudohypoparatyreózy sa prvýkrát potvrdil fenomén narušenej citlivosti tkaniva na hormón produkovaný žľazami s vnútorným vylučovaním alebo ktorý je zavlečený zvonku.
Pseudohypoparatyreóza je patológia genetickej povahy. Spôsobuje ho vrodený syndróm - špecifický bunkový receptor - parathormón-adenylátcykláza, kvôli nemu strácajú periférne tkanivá citlivosť na parathormón.
Kto je najviac náchylný na pseudohypoparatyreózu? Po prvé - deti pacienta, iní príbuzní, pretože choroba má dedičnú autozomálne dominantnú povahu. Ženy trpia Albrightovou chorobou častejšie. Odborníci to vysvetľujú tým, že muži s pseudohypoparatyreózou majú nízku plodnosť a samotná dedičnosť prechádza z otca na syna len v ojedinelých prípadoch.
Ako často sa táto choroba vyskytuje - neexistujú žiadne štatistiky, v lekárskych spisoch je popísaných asi tristo prípadov.
Symptómy pseudohypoparatyreózy
- Poruchy v orgánoch endokrinného systému s pseudohypoparatyreózou spôsobujú aj hlavné príznaky: pacient je zvyčajne podsaditý, nižší ako ostatní v dôsledku skrátených dolných končatín, je obézny, hladina glukózy v krvi je zvýšená, tvár má mesiačiky -tvarovaný tvar;
- U predstaviteľov slabšieho pohlavia je to porušované menštruačný cyklus. Často sú tieto príznaky sprevádzané akromegáliou, gynekomastiou, rôznymi autoimunitnými procesmi, Itsenko-Cushingovým syndrómom a inými ochoreniami. Vyššie u takýchto pacientov a riziko cukrovky a nie cukrovka;
- Tí, ktorí trpia Albrightovou chorobou, sa často sťažujú na problémy so zubami (majú poškodenú sklovinu), pacienti sa obávajú aj zvracania, tonických kŕčov – spontánnych aj vznikajúcich pod vplyvom dráždivých látok, v krvi možno pozorovať moč. Hrozí sivý zákal. Charakterizované mentálnou retardáciou. Deti s týmto ochorením nezvládajú školské učivo, majú zníženú pamäť;
- K výrazným zmenám dochádza aj na osteoartikulárnom systéme – objavuje sa difúzna osteoporóza, vznikajú cysty (hnedé nádory). Pacienti s pseudohypoparatyreózou častejšie trpia zlomeninami alebo deformáciami kostí. Skracujú prvú, štvrtú a piatu záprstnú a metatarzálnu kosť, na rukách a nohách sú známky resorpcie kosti. Okrem toho sa vápnik, ktorý sa uvoľnil z kostí, začína hromadiť v podkožnom tkanive a vytvára kalcifikácie (to sa stáva aj pri myositis ossificans), v oblasti kĺbov, lebečnej dutiny. Kalcifikácie sú prítomné aj vo svaloch, obličkách, myokarde a na stenách veľkých tepien;
- Ďalším znakom Albrightovej choroby je pigmentácia kože. Hnedé škvrny sa šíria po tele (na jednej aj na druhej strane).
Pokiaľ ide o schopnosť pracovať, závisí od závažnosti procesu a účinnosti medikamentózna liečba. Ak je forma ochorenia skrytá a nie sú žiadne zjavné útoky, potom je schopnosť pracovať čiastočne zachovaná. Existuje však množstvo obmedzení - nemožno najmä pracovať s pohyblivými mechanizmami, v doprave, neuropsychickom prepätí, nadmernom fyzická práca. Ak sú časté konvulzívne záchvaty, mentálna retardácia je jasne vyjadrená, výrazné zhoršenie zraku v dôsledku katarakty, potom sa takíto pacienti už považujú za zdravotne postihnutých.
Diagnóza pseudohypoparatyreózy
Príznaky sú dosť špecifické a ak sa začnú objavovať, mali by ste okamžite kontaktovať kvalifikovaného lekára. Je to ranná a správna diagnóza zabezpečuje úspešnosť liečby.
Keďže Albrightova choroba je vrodená, býva pacientovi diagnostikovaná už v detstve (predškolský vek, vek základnej školy). Genetici vyvodzujú závery na základe klinické príznaky používajú sa aj laboratórne a inštrumentálne diagnostické metódy.
Pacient daruje krv najmä na objasnenie diagnózy (u pacientov s pseudohypoparatyreózou je hladina vápnika nízka, fosfor a parathormón vysoká, vysoká a aktivita alkalický fosfát v krvi), moči (u pacienta táto analýza ukáže pokles vylučovaného fosforu a vápnika). Urobia sa aj špeciálne testy, ktoré ukážu pacientovu citlivosť tkanív renálnych tubulov na parathormón. Niekedy je potrebné posúdiť hladinu hydroxyprolínu.
Inštrumentálne metódy zahŕňajú röntgenovú diagnostiku (najinformatívnejšia je pri identifikácii špecifických zmien v kostných a svalových tkanivách).
Liečba pseudohypoparatyreózy
Táto zriedkavá dedičná patológia sa lieči doplnkami vápnika. Dávku vyberá lekár na základe obsahu vápnika potrebného na udržanie homeostázy. A aby sa vápnik bez problémov vstrebal, do terapeutického režimu sú zaradené aj prípravky s vitamínom D. Okrem toho lekár pre pacienta s pseudohypoparatyreózou vypracuje diétu, v ktorej bude obmedzené množstvo produktov s obsahom fluóru. Táto diéta pomôže normalizovať koncentráciu vápnika v krvi a odstrániť príznaky sekundárnej hyperparatyreózy.
Ak je Albrightova choroba sprevádzaná inými endokrinné poruchy, potom môže byť potrebná hormonálna substitučná liečba. Záchvaty sa zmierňujú roztokom chloridu vápenatého alebo glukonátu vápenatého, ktorý sa podáva intravenózne. V prípade sivého zákalu sa do jeho liečby zapája očný lekár, odstraňovanie kognitívnej poruchy pomáha psychológ.
Pri dobre premyslenej schéme terapie je prognóza veľmi priaznivá. Ale všetci, ktorí ochoreli na pseudohypoparatyreózu, by mali pravidelne konzultovať s genetikmi, aby predišli ochoreniu svojich potomkov.
Prevencia pseudohypoparatyreózy
Vzhľadom na to, že ochorenie je dedičné, jediným spôsobom prevencie pre tých, ktorí mali v rodine pseudohypoparatyreózu, sú pravidelné návštevy u lekárov genetika na konzultácie.
- Liečba pseudohypoparatyreózy
- Prevencia pseudohypoparatyreózy
- Ktorých lekárov by ste mali vidieť, ak máte pseudohypoparatyreózu?
Čo je pseudohypoparatyreóza
Pseudohypoparatyreóza(grécky pseudēs false + hypoparatyreóza; synonymum: Albrightova dedičná osteodystrofia, Albrightova choroba) - zriedkavé dedičné ochorenie kostného systému, ktoré napodobňuje hypoparatyreózu a je charakterizované poruchou metabolizmu vápnika a fosforu; často sprevádzané oneskorením duševného a fyzického vývoja.
Čo spôsobuje pseudohypoparatyreózu?
Príčinou pseudohypoparatyreózy je vrodená chyba – necitlivosť periférnych tkanív na pôsobenie PTH.
Patogenéza (čo sa stane?) počas pseudohypoparatyreózy
Predpokladá sa, že základom pseudohypoparatyreózy je geneticky podmienená odolnosť obličiek a kostry voči pôsobeniu parathormónu v dôsledku defektu komplexu špecifického cytoreceptora - parathormónu - adenylátcyklázy, ktorý narúša tvorbu cyklický 3", 5"-AMP v obličkách, ktorý je intracelulárnym mediátorom účinku parathormónu na metabolické procesy. Pseudohypoparatyreóza je geneticky heterogénne ochorenie. U niektorých pacientov je defektný samotný cytoreceptor, ktorý viaže parathormón (pseudohypoparatyreóza typu Ia), u iných je defekt v proteíne viažucom nukleotidy lokalizovaného v lipidovej dvojvrstve bunkovej membrány a funkčne viažuceho receptor na adenylátcyklázu ( pseudohypoparatyreóza typu Ib). Niektorí pacienti majú enzymatický deficit samotnej adenylátcyklázy (pseudohypoparatyreóza typu II). Nedostatok cAMP, ktorý je výsledkom týchto defektov, vedie k narušeniu syntézy špecifických proteínov, ktoré určujú biologický účinok parathormónu. Tým sa stráca citlivosť cieľových orgánov, najmä obličiek, na parathormón. V dôsledku toho sa znižuje vylučovanie fosforu močom, dochádza k hyperfosfatémii a sekundárne sa vyvíja hypokalciémia. Keďže prištítne telieska sú pri pseudohypoparatyreóze intaktné, ako odpoveď na hypokalciémiu, ktorá stimuluje produkciu parathormónu, sekundárneho hyperparatyreóza. Zvýšená tvorba parathormónu nespôsobuje zvýšené vylučovanie fosforu a cAMP močom v dôsledku geneticky podmienenej rezistencie obličkových tubulov na parathormón, ale je sprevádzaná zmenami kostného tkaniva charakteristickými pre hyperparatyreózu, čo poukazuje na tzv. zachovanie normálnej citlivosti osteoklastov na parathormón. Pri pseudohypoparatyreóze je aktivita alkalickej fosfatázy v krvnom sére zvýšená alebo je v normálnom rozmedzí (0,5-1,3 umol anorganický fosfor na 1 ml krvné sérum za 1 h inkubácia pri 37°; definícia podľa Bodanského). Všetky varianty pseudohypoparatyreózy sú dedičné ochorenie, povaha dedičnosti je autozomálne dominantná. Nízka plodnosť mužov trpiacich pseudohypoparatyreózou vysvetľuje vzácnosť jej prenosu z otca na syna; ženy sú choré 2-krát častejšie ako muži.
Zvyčajne sa pri pseudohypoparatyreóze zistí kompenzačná hyperplázia prištítnych teliesok (prítomnosť adenómov v nich nie je typická). V kostnom tkanive sú zaznamenané zmeny typické pre hyperparatyreózu - difúzna osteoporóza, výskyt cýst (tzv. hnedé nádory, nádory obrovských buniek). Vápnik uvoľnený z kostí sa vo forme kalcifikátov ukladá v podkoží, ďalej v obličkách, svaloch, myokarde, stenách veľkých tepien, spojovke oka a po periférii rohovky.
Príznaky pseudohypoparatyreózy
Klinické príznaky pseudohypoparatyreózy sú podobné ako pri idiopatickej hypoparatyreóza. Existujú záchvaty tonických kŕčov, ktoré sa vyskytujú spontánne alebo pod vplyvom akýchkoľvek podnetov. Podkožné kalcifikácie majú tendenciu ulcerovať. Subkutánna osifikácia je často taká výrazná, že napodobňuje myositis ossificans. Charakterizované mentálnou retardáciou, zakrpatením, mesačnou tvárou, obezitou a brachydaktýliou, najmä skrátením prvého, štvrtého a piateho metakarpu a metatarzu. Môžu sa vyskytnúť viaceré exostózy, dyschondroplázia, prejavy sekundárnej hyperparatyreózy vo forme subperiostálnej resorpcie kostí prstov; zmeny v epifýzach kostí sú rovnaké ako pri fibróznej osteodysplázii. Často je zaznamenané vracanie, ako aj hematúria v dôsledku tvorby oxalátových kameňov močové cesty, odhaliť lentikulárnu kataraktu, hypopláziu zubnej skloviny.
U pacientov s pseudohypoparatyreózou, spolu so znížením citlivosti cieľových orgánov na parathormón, rezistenciou na iné hormóny závislé od systému adenylylcyklázy, napríklad pohlavné žľazy na gonadotropné hormóny, štítna žľaza na hormón stimulujúci štítnu žľazu, cieľové orgány pre glukagón a antidiuretický hormón. Existuje zvýšená frekvencia autoimunitné ochorenia A cukrovka pozorovaná hypotyreóza a hypertyreóza.
Rozlišuje sa aj pseudo-pseudohypoparatyreóza, ktorá sa vyznačuje absenciou hypokalcémie, hyperfosfatémie, záchvatov a osteomalácie.
Diagnóza pseudohypoparatyreózy
Diagnóza v typických prípadoch ochorenia je stanovená u detí vo veku 5-10 rokov na základe charakteristického klinického obrazu, mnohopočetných anomálií vo vývoji kostného skeletu, prítomnosti hypokalcémie, hyperfosfatémie, normálnej alebo zvýšenej aktivity alkalickej fosfatázy v krvné sérum, znížené vylučovanie vápnika a fosforu močom, zvýšený obsah parathormónu v krvi. Prítomnosť renálnej tubulárnej rezistencie na parathormón je potvrdená testom založeným na stanovení množstva fosfátu a cAMP vylúčeného močom. Neprítomnosť výrazného zvýšenia obsahu fosfátov a cAMP v moči po podaní parathormónu pacientovi svedčí o rezistencii obličiek voči pôsobeniu parathormónu. U pacientov s idiopatickou a pooperačnou hypoparatyreózou naopak po intravenózne podanie 200 jednotiek parathormónu v moči, obsah fosfátov a cAMP za 4 h zvyšuje 2-10 krát v porovnaní s počiatočnou úrovňou. Vylučovanie hydroxyprolínu močom u neliečených pacientov s pseudohypoparatyreózou je normálne alebo mierne zvýšené a pri hypoparatyreóze je znížené. Röntgenová diagnostika pseudohypoparatyreózy je založená na detekcii špecifických zmien v kostiach a mäkkých tkanivách.
Pseudohypoparatyreoidizmus v kombinácii s hypogonadizmom u žien je potrebné odlíšiť od Sereshevsky - Turnerov syndróm, s ktorým je pseudopseudohypoparatyreóza fenotypovo podobná. Pri Sereshevsky-Turnerovom syndróme neexistuje pohlavný chromatín, namiesto vaječníkov sú vlákna spojivového tkaniva, ktoré nie sú detekované počas rektálneho a ultrazvukové vyšetrenie.
Liečba pseudohypoparatyreózy
Liečba hypokalciémie spočíva v vymenovaní doplnkov vápnika v dávkach dostatočných na udržanie normálnej koncentrácie vápnika v krvi. Veľký význam má terapia vitamínom D. Úvodná dávka sa počíta od 2000 IU / kg telesnej hmotnosti za deň, ale nie viac ako 100 000 IU za deň. Aby sa predišlo predávkovaniu prípravkami vitamínu D, je potrebné sledovať koncentráciu vápnika v krvi každé 3-7 dní počas prvých dvoch týždňov liečby a každý mesiac počas nasledujúcich 2-3 mesiacov. Po dosiahnutí stabilnej koncentrácie vápnika v krvi ho stačí kontrolovať raz za 2-3 mesiace. Môžete použiť kalcitrín, dihydrotachysterol, oxydevit, ako aj iné lieky aktívnych foriem vitamínu D. Diéta s obmedzením fosforu pomáha normalizovať koncentráciu vápnika v krvi a eliminovať príznaky sekundárnej hyperparatyreózy. V prípade nedostatočnosti iných žliaz s vnútornou sekréciou sa vykonáva substitučná liečba vhodnými hormónmi. Liečba parathormónom nie je účinná. Na zmiernenie konvulzívnych záchvatov sa intravenózne podáva 10% roztok chloridu vápenatého alebo glukonátu vápenatého; vnútri - 5-10% roztok chloridu vápenatého, 1 polievková lyžica 3-4 krát denne: glukonát vápenatý, laktát vápenatý - do 10 G o deň.
Prognóza racionálnej terapie je priaznivá. Vzhľadom na dedičnú povahu pseudohypoparatyreózy je potrebné lekárske genetické poradenstvo týkajúce sa možnosti pseudohypoparatyreózy u potomkov.
Pseudohypoparatyreóza
Čo je Pseudohypoparatyreóza?
Pseudohypoparatyreóza h(Grécke pseudē je falošné + hypoparatyreóza; synonymum: Albrightova dedičná osteodystrofia, Albrightova choroba) je zriedkavé dedičné ochorenie kostrového systému, ktoré napodobňuje hypoparatyreózu a vyznačuje sa poruchou metabolizmu vápnika a fosforu; často sprevádzané oneskorením duševného a fyzického vývoja.
Čo vyvoláva / Príčiny pseudohypoparatyreózy:
Príčinou pseudohypoparatyreózy je vrodená chyba – necitlivosť periférnych tkanív na pôsobenie PTH.
Patogenéza (čo sa stane?) počas pseudohypoparatyreózy:
Predpokladá sa, že základom pseudohypoparatyreózy je geneticky podmienená odolnosť obličiek a kostry voči pôsobeniu parathormónu v dôsledku defektu komplexu špecifického cytoreceptora - parathormónu - adenylátcyklázy, ktorý narúša tvorbu cyklický 3", 5"-AMP v obličkách, ktorý je intracelulárnym mediátorom účinku parathormónu na metabolické procesy. Pseudohypoparatyreóza je geneticky heterogénne ochorenie. U niektorých pacientov je defektný samotný cytoreceptor, ktorý viaže parathormón (pseudohypoparatyreóza typu Ia), u iných je defekt v proteíne viažucom nukleotidy lokalizovaného v lipidovej dvojvrstve bunkovej membrány a funkčne viažuceho receptor na adenylátcyklázu ( pseudohypoparatyreóza typu Ib). Niektorí pacienti majú enzymatický deficit samotnej adenylátcyklázy (pseudohypoparatyreóza typu II). Nedostatok cAMP, ktorý je výsledkom týchto defektov, vedie k narušeniu syntézy špecifických proteínov, ktoré určujú biologický účinok parathormónu. Tým sa stráca citlivosť cieľových orgánov, najmä obličiek, na parathormón. V dôsledku toho sa znižuje vylučovanie fosforu močom, dochádza k hyperfosfatémii a sekundárne sa vyvíja hypokalciémia. Keďže prištítne telieska sú pri pseudohypoparatyreóze intaktné, ako odpoveď na hypokalciémiu, ktorá stimuluje produkciu parathormónu, sekundárneho hyperparatyreóza. Zvýšená tvorba parathormónu nespôsobuje zvýšené vylučovanie fosforu a cAMP močom v dôsledku geneticky podmienenej rezistencie obličkových tubulov na parathormón, ale je sprevádzaná zmenami kostného tkaniva charakteristickými pre hyperparatyreózu, čo poukazuje na tzv. zachovanie normálnej citlivosti osteoklastov na parathormón. Pri pseudohypoparatyreóze je aktivita alkalickej fosfatázy v krvnom sére zvýšená alebo je v normálnom rozmedzí (0,5-1,3 umol anorganický fosfor na 1 ml krvné sérum za 1 h inkubácia pri 37°; definícia podľa Bodanského). Všetky varianty pseudohypoparatyreózy sú dedičné ochorenie, povaha dedičnosti je autozomálne dominantná. Nízka plodnosť mužov trpiacich pseudohypoparatyreózou vysvetľuje vzácnosť jej prenosu z otca na syna; ženy sú choré 2-krát častejšie ako muži.
Zvyčajne sa pri pseudohypoparatyreóze zistí kompenzačná hyperplázia prištítnych teliesok (prítomnosť adenómov v nich nie je typická). V kostnom tkanive sú zaznamenané zmeny typické pre hyperparatyreózu - difúzna osteoporóza, výskyt cýst (tzv. hnedé nádory, nádory obrovských buniek). Vápnik uvoľnený z kostí sa vo forme kalcifikátov ukladá v podkoží, ďalej v obličkách, svaloch, myokarde, stenách veľkých tepien, spojovke oka a po periférii rohovky.
Symptómy pseudohypoparatyreózy:
Klinické príznaky pseudohypoparatyreózy sú podobné ako pri idiopatickej hypoparatyreóza. Existujú záchvaty tonických kŕčov, ktoré sa vyskytujú spontánne alebo pod vplyvom akýchkoľvek podnetov. Podkožné kalcifikácie majú tendenciu ulcerovať. Subkutánna osifikácia je často taká výrazná, že napodobňuje myositis ossificans. Charakterizované mentálnou retardáciou, zakrpatením, mesačnou tvárou, obezitou a brachydaktýliou, najmä skrátením prvého, štvrtého a piateho metakarpu a metatarzu. Môžu sa vyskytnúť viaceré exostózy, dyschondroplázia, prejavy sekundárnej hyperparatyreózy vo forme subperiostálnej resorpcie kostí prstov; zmeny v epifýzach kostí sú rovnaké ako pri fibróznej osteodysplázii. Často sa zaznamenáva vracanie, ako aj hematúria v dôsledku tvorby oxalátových kameňov v močovom trakte, zisťuje sa lentikulárna katarakta a hypoplázia zubnej skloviny.
U pacientov so spseudohypoparatyreózou, spolu so znížením citlivosti cieľových orgánov na parathormón, rezistenciou na iné hormóny závislé od systému adenylátcyklázy, ako sú pohlavné žľazy na gonadotropné hormóny, štítna žľaza na hormón stimulujúci štítnu žľazu, cieľové orgány na glukagón a antidiuretický hormón. Existuje zvýšená frekvencia autoimunitné ochorenia A cukrovka pozorovaná hypotyreóza a hypertyreóza.
Rozlišuje sa aj pseudo-pseudohypoparatyreóza, ktorá sa vyznačuje absenciou hypokalcémie, hyperfosfatémie, záchvatov a osteomalácie.
Diagnóza pseudohypoparatyreózy:
Diagnóza v typických prípadoch ochorenia je stanovená u detí vo veku 5-10 rokov na základe charakteristického klinického obrazu, mnohopočetných anomálií vo vývoji kostného skeletu, prítomnosti hypokalcémie, hyperfosfatémie, normálnej alebo zvýšenej aktivity alkalickej fosfatázy v krvné sérum, znížené vylučovanie vápnika a fosforu močom, zvýšený obsah parathormónu v krvi. Prítomnosť renálnej tubulárnej rezistencie na parathormón je potvrdená testom založeným na stanovení množstva fosfátu a cAMP vylúčeného močom. Neprítomnosť výrazného zvýšenia obsahu fosfátov a cAMP v moči po podaní parathormónu pacientovi svedčí o rezistencii obličiek voči pôsobeniu parathormónu. U pacientov s idiopatickou a pooperačnou hypoparatyreózou je naopak po intravenóznom podaní 200 jednotiek parathormónu v moči obsah fosfátov a cAMP na 4. h zvyšuje 2-10 krát v porovnaní s počiatočnou úrovňou. Vylučovanie hydroxyprolínu močom u neliečených pacientov s pseudohypoparatyreózou je normálne alebo mierne zvýšené a pri hypoparatyreóze je znížené. Röntgenová diagnostika pseudohypoparatyreózy je založená na detekcii špecifických zmien v kostiach a mäkkých tkanivách.
Pseudohypoparatyreoidizmus v kombinácii s hypogonadizmom u žien je potrebné odlíšiť od Shereshevsky-Turnerov syndróm, s ktorým je pseudopseudohypoparatyreóza fenotypovo podobná. Pri Shereshevsky-Turnerovom syndróme neexistuje pohlavný chromatín, namiesto vaječníkov sú vlákna spojivového tkaniva, ktoré nie sú detegované rektálnym a ultrazvukovým vyšetrením.
Liečba pseudohypoparatyreózy:
Liečba hypokalciémie spočíva v vymenovaní doplnkov vápnika v dávkach dostatočných na udržanie normálnej koncentrácie vápnika v krvi. Veľký význam má terapia vitamínom D. Úvodná dávka sa počíta od 2000 IU / kg telesnej hmotnosti za deň, ale nie viac ako 100 000 IU za deň. Aby sa predišlo predávkovaniu prípravkami vitamínu D, je potrebné sledovať koncentráciu vápnika v krvi každé 3-7 dní počas prvých dvoch týždňov liečby a každý mesiac počas nasledujúcich 2-3 mesiacov. Po dosiahnutí stabilnej koncentrácie vápnika v krvi ho stačí kontrolovať raz za 2-3 mesiace. Môžete použiť kalcitrín, dihydrotachysterol, oxydevit, ako aj iné lieky aktívnych foriem vitamínu D. Diéta s obmedzením fosforu pomáha normalizovať koncentráciu vápnika v krvi a eliminovať príznaky sekundárnej hyperparatyreózy. V prípade nedostatočnosti iných žliaz s vnútornou sekréciou sa vykonáva substitučná liečba vhodnými hormónmi. Liečba parathormónom nie je účinná. Na zmiernenie konvulzívnych záchvatov sa intravenózne podáva 10% roztok chloridu vápenatého alebo glukonátu vápenatého; vnútri - 5-10% roztok chloridu vápenatého, 1 polievková lyžica 3-4 krát denne: glukonát vápenatý, laktát vápenatý - do 10 G o deň.
Prognóza racionálnej terapie je priaznivá. Vzhľadom na dedičnú povahu pseudohypoparatyreózy je potrebné lekárske genetické poradenstvo týkajúce sa možnosti pseudohypoparatyreózy u potomkov.
Prevencia pseudohypoparatyreózy:
Ktorých lekárov by ste mali kontaktovať, ak máte pseudohypoparatyreózu:
Máte z niečoho obavy? Chcete vedieť podrobnejšie informácie o Pseudohypoparatyreóze, jej príčinách, príznakoch, spôsoboch liečby a prevencie, priebehu ochorenia a strave po ňom? Alebo potrebujete kontrolu? Môžeš objednať sa k lekárovi- poliklinika eurlaboratórium vždy k vašim službám! Najlepší lekári skúmať ťa, študovať vonkajšie znaky a pomôcť identifikovať ochorenie podľa príznakov, poradiť a poskytnúť potreboval pomoc a stanoviť diagnózu. môžete tiež zavolajte lekára domov. Poliklinika eurlaboratórium otvorené pre vás 24 hodín denne.
Ako kontaktovať kliniku:
Telefón našej kliniky v Kyjeve: (+38 044) 206-20-00 (multikanál). Sekretárka kliniky vám vyberie vhodný deň a hodinu návštevy lekára. Naše súradnice a smer sú uvedené. Pozrite sa na ňu podrobnejšie o všetkých službách kliniky.
(+38 044) 206-20-00
Ak ste v minulosti vykonali nejaký výskum, ich výsledky určite zoberte na konzultáciu s lekárom. Ak štúdie nie sú ukončené, urobíme všetko potrebné na našej klinike alebo s kolegami na iných klinikách.
ty? Musíte byť veľmi opatrní na svoje celkové zdravie. Ľudia nevenujú dostatočnú pozornosť symptómy ochorenia a neuvedomujú si, že tieto choroby môžu byť život ohrozujúce. Je veľa chorôb, ktoré sa na našom tele najskôr neprejavia, no nakoniec sa ukáže, že na ich liečbu je už, žiaľ, neskoro. Každá choroba má svoje špecifické príznaky, charakteristické vonkajšie prejavy- tzv symptómy ochorenia. Identifikácia symptómov je prvým krokom k diagnostike chorôb vo všeobecnosti. Ak to chcete urobiť, musíte to urobiť niekoľkokrát do roka byť vyšetrený lekárom nielen zabrániť hrozná choroba ale aj na udržanie zdravej mysle v tele a celkovom tele.
Ak chcete lekárovi položiť otázku, využite sekciu online poradne, možno tam nájdete odpovede na svoje otázky a čítate tipy na starostlivosť o seba. Ak vás zaujímajú recenzie o klinikách a lekároch, skúste si potrebné informácie nájsť v sekcii. Zaregistrujte sa aj na lekárskom portáli eurlaboratórium byť neustále aktuálny najnovšie správy a aktualizácie informácií na stránke, ktoré vám budú automaticky zasielané poštou.
Ďalšie choroby zo skupiny Choroby endokrinného systému, poruchy príjmu potravy a poruchy látkovej výmeny:
| Addisonova kríza (akútna adrenálna insuficiencia) |
| adenóm prsníka |
| Adiposogenitálna dystrofia (Perchkrantz-Babinski-Fröhlichova choroba) |
| Adrenogenitálny syndróm |
| Akromegália |
| Alimentárne šialenstvo (alimentárna dystrofia) |
| Alkalóza |
| Alkaptonúria |
| Amyloidóza (degenerácia amyloidu) |
| Amyloidóza žalúdka |
| Črevná amyloidóza |
| Amyloidóza pankreatických ostrovčekov |
| Amyloidóza pečene |
| Amyloidóza pažeráka |
| Acidóza |
| Proteínová energetická podvýživa |
| I-bunkové ochorenie (mukolipidóza typu II) |
| Wilsonova-Konovalovova choroba (hepatocerebrálna dystrofia) |
| Gaucherova choroba (glukocerebrozidová lipidóza, glukocerebrosidóza) |
| Itsenko-Cushingova choroba |
| Krabbeho choroba (globoidná bunková leukodystrofia) |
| Niemann-Pickova choroba (sfingomyelinóza) |
| Fabryho choroba |
| Gangliosidóza GM1 typu I |
| Gangliosidóza GM1 typu II |
| Gangliosidóza GM1 typ III |
| Gangliosidóza GM2 |
| GM2 gangliozidóza typu I (Tay-Sachsova amaurotická idiocia, Tay-Sachsova choroba) |
| Gangliosidóza GM2 typu II (Sandhoffova choroba, Sandhoffova amaurotická idiocia) |
| Gangliosidóza GM2 juvenilná |
| Gigantizmus |
| Hyperaldosteronizmus |
| Sekundárny hyperaldosteronizmus |
| Primárny hyperaldosteronizmus (Connov syndróm) |
| Hypervitaminóza D |
| Hypervitaminóza A |
| Hypervitaminóza E |
| Hypervolémia |
| Hyperglykemická (diabetická) kóma |
| Hyperkaliémia |
| Hyperkalcémia |
| Hyperlipoproteinémia typu I |
| Hyperlipoproteinémia typu II |
| Hyperlipoproteinémia typu III |
| Hyperlipoproteinémia IV. typu |
| Hyperlipoproteinémia typu V |
| Hyperosmolárna kóma |
| Sekundárna hyperparatyreóza |
| Primárna hyperparatyreóza |
| Hyperplázia týmusu (brzlík) |
| Hyperprolaktinémia |
| hyperfunkcia semenníkov |
| Hypercholesterolémia |
| hypovolémia |
| Hypoglykemická kóma |
| hypogonadizmus |
| Hypogonadizmus hyperprolaktinémia |
| Izolovaný hypogonadizmus (idiopatický) |
| Hypogonadizmus primárne vrodený (anorchizmus) |
| Hypogonadizmus, primárne získaný |
| hypokaliémia |
| Hypoparatyreóza |
| hypopituitarizmus |
| Hypotyreóza |
| Glykogenóza typu 0 (aglykogenóza) |
| Glykogenóza typu I (Girkeova choroba) |
| Glykogenóza typu II (Pompeho choroba) |
| Glykogenóza typu III (ochorenie osýpok, Forbesova choroba, limitná dextrinóza) |
| Glykogenóza IV. typu (Andersenova choroba, amylopektinóza, difúzna glykogenóza s cirhózou pečene) |
| Glykogenóza typu IX (Hagova choroba) |
| Glykogenóza typu V (McArdleova choroba, nedostatok myofosforylázy) |
| Glykogenóza typu VI (jej choroba, nedostatok hepatofosforylázy) |
| Glykogenóza typu VII (Taruiova choroba, nedostatok myofosfofruktokinázy) |
| Glykogenóza typu VIII (Thomsonova choroba) |
| Glykogenóza typu XI |
| Glykogenóza typu X |
| Nedostatok (nedostatok) vanádu |
| Nedostatok (nedostatok) horčíka |
| Nedostatok (nedostatok) mangánu |
| Nedostatok (nedostatok) medi |
| Nedostatok (nedostatok) molybdénu |
| Nedostatok (nedostatok) chrómu |
| nedostatok železa |
| Nedostatok vápnika (alimentárny nedostatok vápnika) |
| Nedostatok zinku (alimentárny nedostatok zinku) |
| diabetická ketoacidotická kóma |
| Dysfunkcia vaječníkov |
| Difúzna (endemická) struma |
| Oneskorená puberta |
| Nadbytok estrogénu |
| Involúcia mliečnych žliaz |
| nanizmus (nízky vzrast) |
| Kwashiorkor |
| Cystická mastopatia |
| xantínúria |
| Laktickú kómu |
| Leucinóza (choroba javorového sirupu) |
| Lipidózy |
| Farberova lipogranulomatóza |
| Lipodystrofia (degenerácia tukov) |
| Generalizovaná vrodená lipodystrofia (Sape-Lawrenceov syndróm) |
| Hypermuskulárna lipodystrofia |
| Lipodystrofia po injekcii |
| Lipodystrofia progresívna segmentálna |
| Lipomatóza |
| Lipomatóza bolestivá |
| Metachromatická leukodystrofia |
| Myxedémová kóma |
